不同液相色谱-质谱联用法分析贝类样品中神经毒素β-N-甲氨基-L-丙氨酸的比较
2021-03-03王超邱江兵宋甲亮李爱峰
王超,邱江兵,宋甲亮,李爱峰,2*
(1. 中国海洋大学环境科学与工程学院; 2. 海洋环境与生态教育部重点实验室: 山东 青岛 266100)
β-N-甲氨基-L-丙氨酸(β-N-methylamino-L-alanine, BMAA)是一种具有神经毒性的非蛋白质氨基酸[1],含有一个羧基和一个氨基,具有甲氨基侧链(图1),其化学性质稳定,易溶于水。目前已在自然界中发现了多种BMAA的同分异构体,如常见的β-氨基-N-甲基丙氨酸(β-amino-N-methylalanine, BAMA)、2,4-二氨基丁酸(2,4-diaminobutyric acid, DAB)、N-2-氨乙基甘氨酸[N-(2-aminoethyl)glycine, AEG]等,均具有一定的神经毒性。据报道,水环境中分布的蓝藻[2-5]、硅藻[6-10]和甲藻[11]样品均曾检出BMAA,且发现了BMAA沿食物链传递的生物放大现象[12-14]。研究表明BMAA对运动神经元细胞具有毒性作用[15-19],且在关岛及关岛以外地区的神经退行性疾病相关的患者脑组织中检出BMAA[20-21],因此研究推测BMAA与全球神经退行性疾病发病率逐渐升高的现象有关。
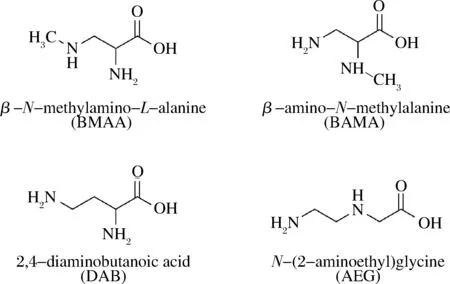
图1 BMAA、BAMA、DAB和AEG的化学结构Fig.1 Chemical structures of BMAA, BAMA, DAB and AEG
由于BMAA属于水溶性的小分子化合物,且同分异构体化合物较多,对于BMAA的定性和定量分析提出了挑战。目前用于BMAA的分析方法包括毛细管电泳法(CE)[3]、气相色谱-质谱联用法(GC-MS)[5]、液相色谱-荧光检测法(LC-FD)[2]和液相色谱-质谱联用法(LC-MS)[20]等,但目前有关这些方法分析不同生物基质样品中BMAA毒素的准确度尚缺少实验室间的互校验证,对某些环境样品中BMAA的定量结果存在较大偏差[22]。其中液相色谱-串联质谱法通过保留时间、母离子质荷比、碎片离子的质荷比及其丰度比值作为定性依据,对BMAA分析具有很高的选择性和灵敏度,已成为分析BMAA的最常用的方法,可分为柱前衍生法和直接分析法。
柱前衍生法主要是借鉴氨基酸的液相色谱分析方法,采用6-氨基喹啉基-N-羟基琥珀酰亚氨基甲酸酯(6-aminoquinolyl-N-hydroxysuccinimidyl carbamate, AQC)作为BMAA衍生剂[23],生成分子量和疏水性更高的喹啉化合物,通过C18反向色谱柱实现与其他化合物的分离。直接分析法是采用亲水交互作用(hydrophilic liquid interaction chromatography, HILIC)色谱柱,使得亲水性的BMAA及其同分异构体化合物具有不同的保留能力,实现柱上分离。后来研究发现BMAA及其同分异构体BAMA的分离度较低,Foss等[24]建立的HILIC-MS/MS方法能够实现二者的相对分离,但也不能将BMAA与BAMA完全分离。综合考虑直接分析方法不需要样品的衍生化过程,节约了样品制备的成本和时间,因此在BMAA的环境样品分析中得到了广泛的应用[25-28]。
随着对环境样品中BMAA的深入分析,发现世界多个海区采集的软体动物样品普遍含有BMAA[28-31],在中国沿海城市采集的软体动物样品中也普遍检出BMAA[32-33]。本研究选择中国沿海常见的贝类——扁玉螺(Neveritadidyma)、脉红螺(Rapanavenosa)、栉江珧(Atrinapectinata)和菲律宾蛤仔(Ruditapesphilippinarum)为研究对象,比较了LC-MS/MS直接分析法和AQC柱前衍生法定性和定量分析BMAA的优缺点,以期为生物样品中BMAA毒素精准分析技术的研发提供参考。
1 实验部分
1.1 仪器与试剂
1290高效液相色谱和三重四极杆质谱6430(美国安捷伦公司); Ultimate 3000高效液相色谱(美国Thermo Fisher Scientific公司)和Qtrap 4500三重四极杆/复合线性离子阱质谱(美国AB Sciex公司);超声清洗仪(昆山市超声仪器有限公司);超纯水机(美国Millipore公司)。
标准毒素BMAA(≥97% NMR级,美国Sigma公司)、标准毒素DAB(≥98% TLC级,美国Sigma公司);标准毒素AEG(加拿大TRC公司);甲酸、甲醇和乙腈(色谱级,德国Merck公司);三氯乙酸(分析级,美国Sigma公司);0.22 μm水系滤膜(天津津腾公司)。
1.2 样品处理与衍生方法
1.2.1 扁玉螺样品的制备方法
目前有关生物样品中BMAA毒素的分析一般同时考虑3个形态的分布(图2):游离态是指提取溶液中游离存在的BMAA毒素分子;总溶解态是指提取溶液中分散的游离态毒素分子及其嵌入到短的肽链中的毒素分子,常规的离心条件不能使其沉淀,包括游离态和溶解结合态两部分;沉淀结合态是指被错误地编码进入蛋白质的BMAA毒素分子。本研究对采集的3批扁玉螺样品中的3种形态的BMAA毒素进行了分析。

图2 BMAA毒素的3种分布形态Fig.2 Different forms of BMAA in an aqueous extract
扁玉螺是已知的一种BMAA毒素的典型携带生物,本研究将采自荣成、大连和青岛的3批扁玉螺样品分别标记为S01、S02和S03,作为阳性样品。取扁玉螺的全部软体组织搅碎、冷冻干燥,然后使用研钵将其充分研磨粉碎。采用0.1 mol/L TCA为提取溶剂,使用2个不同的提取比例制备样品。首先,称取0.5 g扁玉螺粉末样品,加入3 mL TCA,涡旋震荡1 min,离心10 min(6 577 × g),重复提取3次;使用0.1 mol/L TCA将混合后的提取溶液定容至10 mL,提取比例为20∶1(10 mL /0.5 g干重)。其次,称量0.2 g扁玉螺粉末样品,采用上述相同的方法重复提取3次,提取比例为50∶1(10 mL /0.2 g干重)。
游离态BMAA:取1 mL离心后的上清液过0.22 μm滤膜,作为游离态BMAA毒素样品,用于LC-MS/MS分析。
总溶解态BMAA:取1 mL离心后的上清液在55 ℃条件下用氮气吹干,然后用1 mL 6 mol/L HCl 溶液重新溶解,涡旋震荡混匀,110 ℃条件下水解24 h。水解完成后,冷却至室温,氮气吹干,重新溶解于1 mL 20 mmol/L HCl中,过0.22 μm滤膜用于LC-MS/MS分析。该样品包括游离态和溶解结合态BMAA毒素。
沉淀结合态BMAA:向提取游离态BMAA毒素后剩余的蛋白质沉淀中加入3 mL 6 mol/L HCl,涡旋震荡混匀后转移至4 mL玻璃瓶中,在110 ℃高温条件下水解24 h。待冷却至室温后,在55 ℃条件下用氮气吹干,然后加入3 mL 20 mmol/L HCl 溶液重新悬浮,离心10 min(6 577 × g)后将上清液转移至10 mL容量瓶中,用20 mmol/L HCl 定容至10 mL。最后取1 mL提取液过0.22 μm滤膜用于毒素分析。
BMAA毒素的衍生化步骤:取1 mL乙腈溶解AQC粉末试剂,涡旋混匀后,55 ℃条件下加热促溶,得到AQC衍生剂。取20 μL BMAA样品提取液,依次加入60 μL AQC缓冲溶液和20 μL AQC衍生剂,立即涡旋混匀;然后放在55 ℃条件下加热反应10 min,再次涡旋震荡,等待LC-MS/MS分析。
1.2.2 其他贝类样品的制备方法
鉴于前文在扁玉螺样品中未检出蛋白结合态BMAA毒素,本研究分析其他贝类样品时只检测了新鲜的贝肉组织中总溶解态BMAA的含量,未使用冷冻干燥方法处理,但使用固相萃取柱(solid phase extraction, SPE)对样品进行了纯化。与1.2.1部分样品制备方法的不同之处如下:
称取0.5 g匀浆的新鲜贝肉样品置于10 mL离心管中,加入3 mL 0.1 mol/L TCA,涡旋震荡1 min, 再次进行组织匀浆,充分打碎;然后涡旋震荡1 min, 离心10 min(6 577×g),取上清液转移至10 mL容量瓶,重复上述步骤3次,合并上清液并用0.1 mol/L TCA定容至10 mL。取1 mL上清液在55 ℃条件下用氮气吹干,然后用1 mL 6 mol/L HCl 溶液重新溶解,涡旋震荡混匀,110 ℃条件下水解24 h。水解完成后,冷却至室温,氮气吹干,重新溶解于1 mL 20 mmol/L HCl中,使用Oasis MCX(3 cc, 60 mg)阳离子交换SPE小柱进行纯化,具体步骤如下:先后使用3 mL甲醇和2 mL超纯水,活化和平衡固相萃取柱;然后加入1 mL BMAA毒素提取液;待样品全部流经固相萃取柱后,依次加入2 mL 0.1 mol/L HCl和2 mL甲醇,洗涤提取液中的杂质;最后使用 3 mL 5%氨水溶液(NH3·H2O)进行洗脱并收集洗脱液,在55 ℃条件下用氮气吹干,再用1 mL 20 mmol/L HCl重新溶解残余物,并经0.22 μm水系滤膜过滤后待分析,作为总溶解态毒素样品。
这些贝类样品的总溶解态BMAA毒素的提取溶液均采用1.2.1部分相同的AQC衍生法进行柱前衍生。
1.3 色谱和质谱条件
1.3.1 LC-MS/MS直接分析法
(1)扁玉螺样品分析的LC-MS/MS条件
色谱柱为TSK Gel Amide-80(250 mm × 2 mm, 5 μm, Tosoh Corporation),进样量为5 μL,流速为350 μL/min,柱温设置为40 ℃,流动相A为含有50 mmol/L甲酸的水溶液,流动相B为含有50 mmol/L甲酸的95%乙腈溶液。流动相采用线性梯度洗脱:0~15 min,流动相B从90%降低至60%;15~19 min,流动相B保持在60%;在19.01 min将流动相B的比例降至55%并维持至27 min。Agilent 6430质谱的电喷雾离子化电压为5 500 V,离子源温度为450 ℃,使用氮气作为雾化和气帘气体。目标化合物检测采用选择反应监测(SRM)模式,具体参数见表1。
(2)其他贝类样品分析的LC-MS/MS条件
色谱柱为SeQuant ZIC HILIC色谱柱(150 mm×2.1 mm, 5 μm,Merck KGaA),进样量为5 μL,流速为350 μL/min,柱温设置为30 ℃,流动相A为含有0.1%甲酸的水溶液,流动相B为含有0.1%甲酸的乙腈溶液。采用梯度洗脱:0~19 min,95%~ 60%流动相B;19~25 min,60%~40%流动相B;25~27 min,40%~95%流动相B,保持3 min。AB Sciex Qtrap 4500质谱的电喷雾电压5 500 V,雾化气压力50 psi,辅助气压力50 psi,雾化温度350 ℃,气帘气压力40 psi,入口电压10 V,碰撞室射出电压12 V。目标化合物检测采用选择反应监测(SRM)模式,具体参数见表1。
1.3.2 LC-MS/MS衍生分析法
(1)扁玉螺样品分析的LC-MS/MS条件
色谱柱为Phenomenex Kinetex C18色谱柱(100 mm×2.1 mm, 1.7 μm, 100Å),进样量为5 μL,流速为350 μL/min,柱温设置为55 ℃,流动相A为含有20 mmol/L甲酸铵的水溶液(冰醋酸调节pH=5),流动相B为甲醇。流动相采用线性梯度洗脱的方法:0~2.5 min,流动相A从72%降低至62%,然后保持1 min;3.5~4 min,流动相A降低至15%;然后在0.25 min内流动相A降低至10%,并保持1 min;最后在0.2 min内流动相A恢复至72%,并保持7 min。质谱Agilent 6430的条件参数见1.3.1部分,SRM模式检测BMAA及其同系物的具体参数见表2。
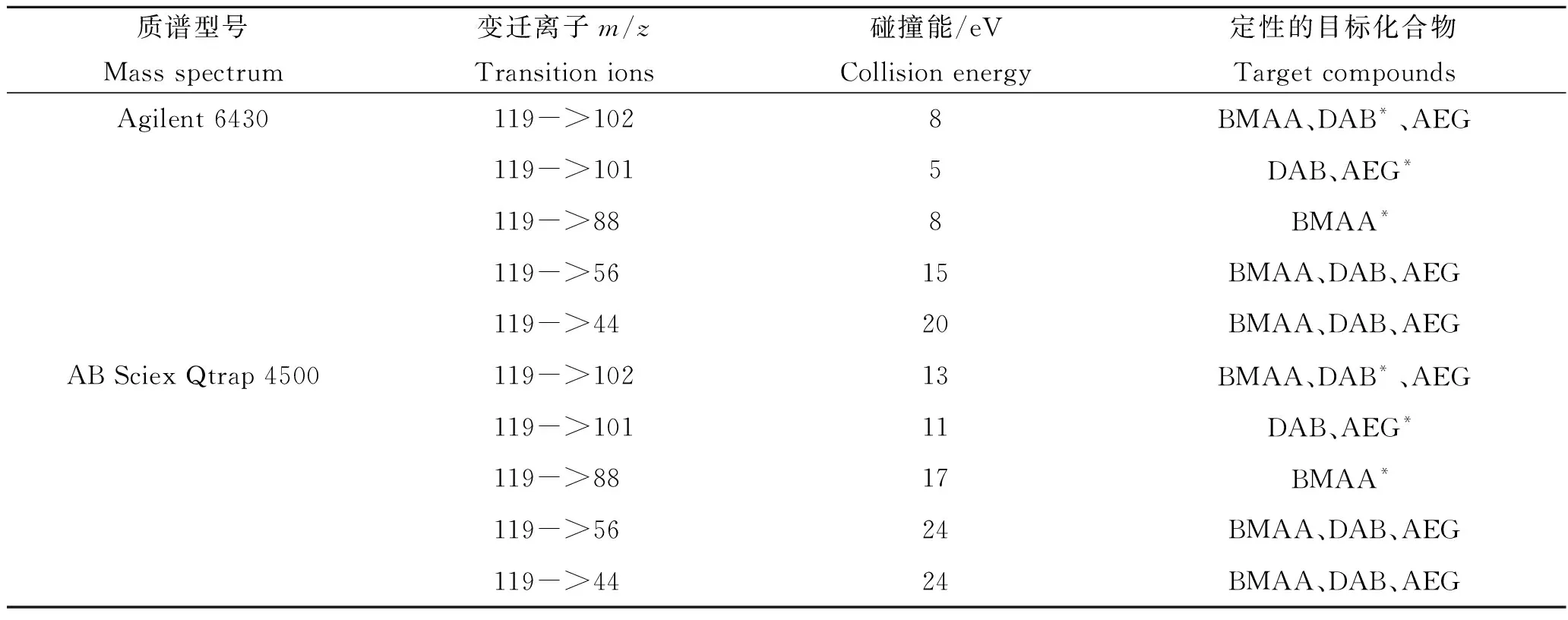
表1 选择反应监测模式检测BMAA及其同分异构体化合物的质谱参数Tab.1 LC-MS/MS parameters of the selective reaction monitoring mode for BMAA and its isomers
(2)其他生物样品分析的LC-MS/MS条件
色谱柱采用相同的Phenomenex Kinetex C18色谱柱,进样量为5 μL,流速为350 μL/min,柱温设置为65oC,流动相A和B的成分与上述方法相同,但梯度洗脱条件有所改变。0~2.5 min,28%~38% B,保持1 min;3.5~4 min,38%~85% B;4~4.25 min,85%~90% B,保持1 min;5.25~5.45 min,90%~28% B,保持1.55 min。质谱AB Sciex Qtrap 4500的条件见1.3.1部分,SRM模式检测BMAA及其同系物的具体参数见表2。
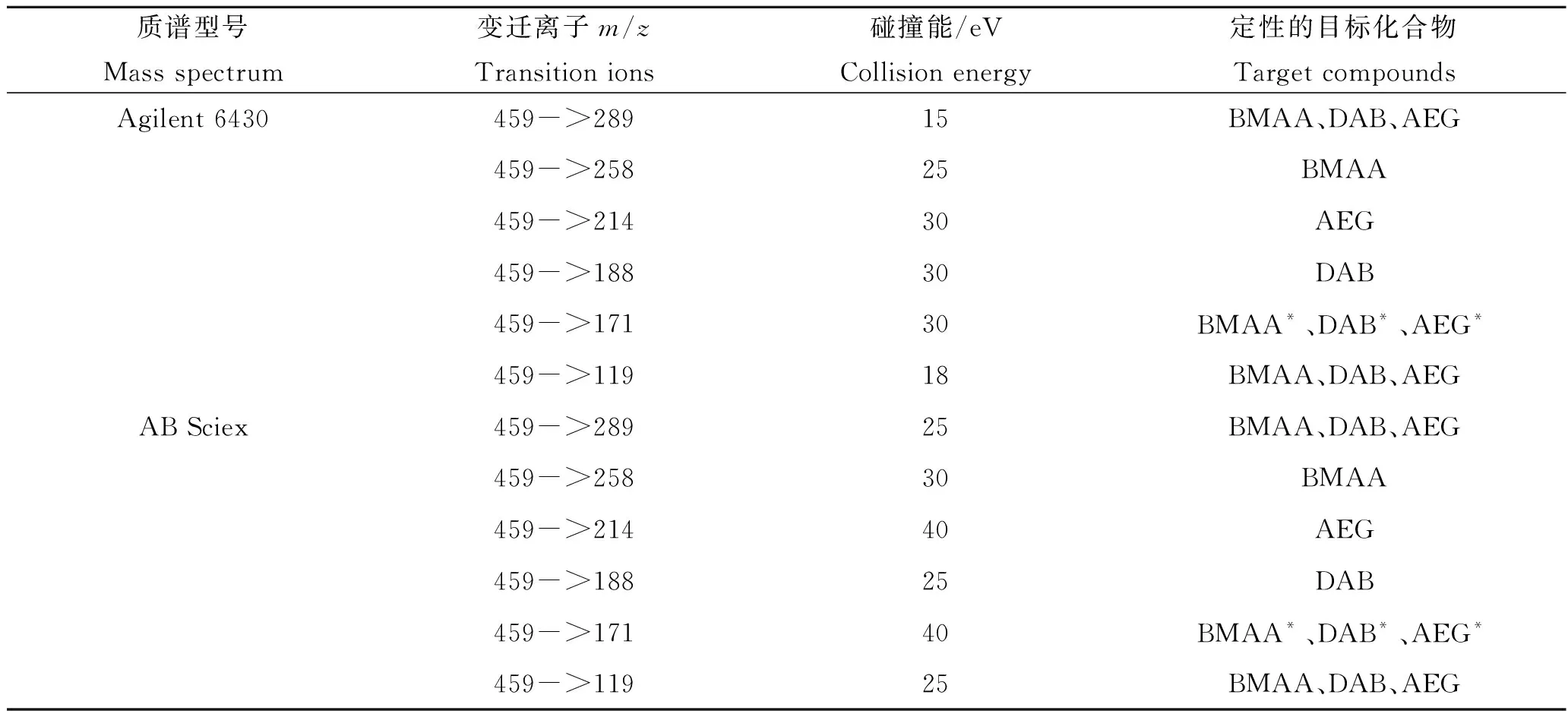
表2 选择反应监测模式检测BMAA及其同系物的AQC衍生物的质谱参数Tab.2 LC-MS/MS parameters of the selective reaction monitoring mode for the AQC derivatives of BMAA and its isomers
2 结果与讨论
2.1 扁玉螺样品中BMAA及其异构体化合物的分析
亲水交互作用色谱柱TSK Gel Amide-80分析扁玉螺样品中BMAA及其同系物的色谱图与前期研究的结果相似[32],添加到样品中的BMAA、DAB和AEG 3种毒素在13~17 min范围内可以实现基线分离,且BMAA毒素具有很好的峰形,标准毒素、加标样品和实际样品中BMAA毒素的3种变迁离子(119->102、56和44)峰面积之间的比值相似,变迁离子119->88峰面积的相对比值几乎相同[32]。鉴于DAB和AEG在所用质谱系统中均不具有变迁离子119->88,建议使用该变迁离子的色谱峰面积计算BMAA毒素的浓度。LC-MS/MS分析AQC衍生后的扁玉螺样品的典型色谱图如图3所示。从图中可以看出BMAA和DAB的AQC衍生物在C18色谱柱上实现了基线分离,但在保留时间2.1 min位置出现一个很小的色谱峰,根据其相对BMAA的保留时间,参考前期已报道的BAMA与BMAA在C18色谱柱上的分离度[34],可推测该痕量物质应该是BMAA的同分异构体化合物BAMA。由此,可以推断在使用TSK Gel Amide-80 HILIC色谱柱分析扁玉螺的过程中,BMAA的色谱峰中掺杂痕量的BAMA。
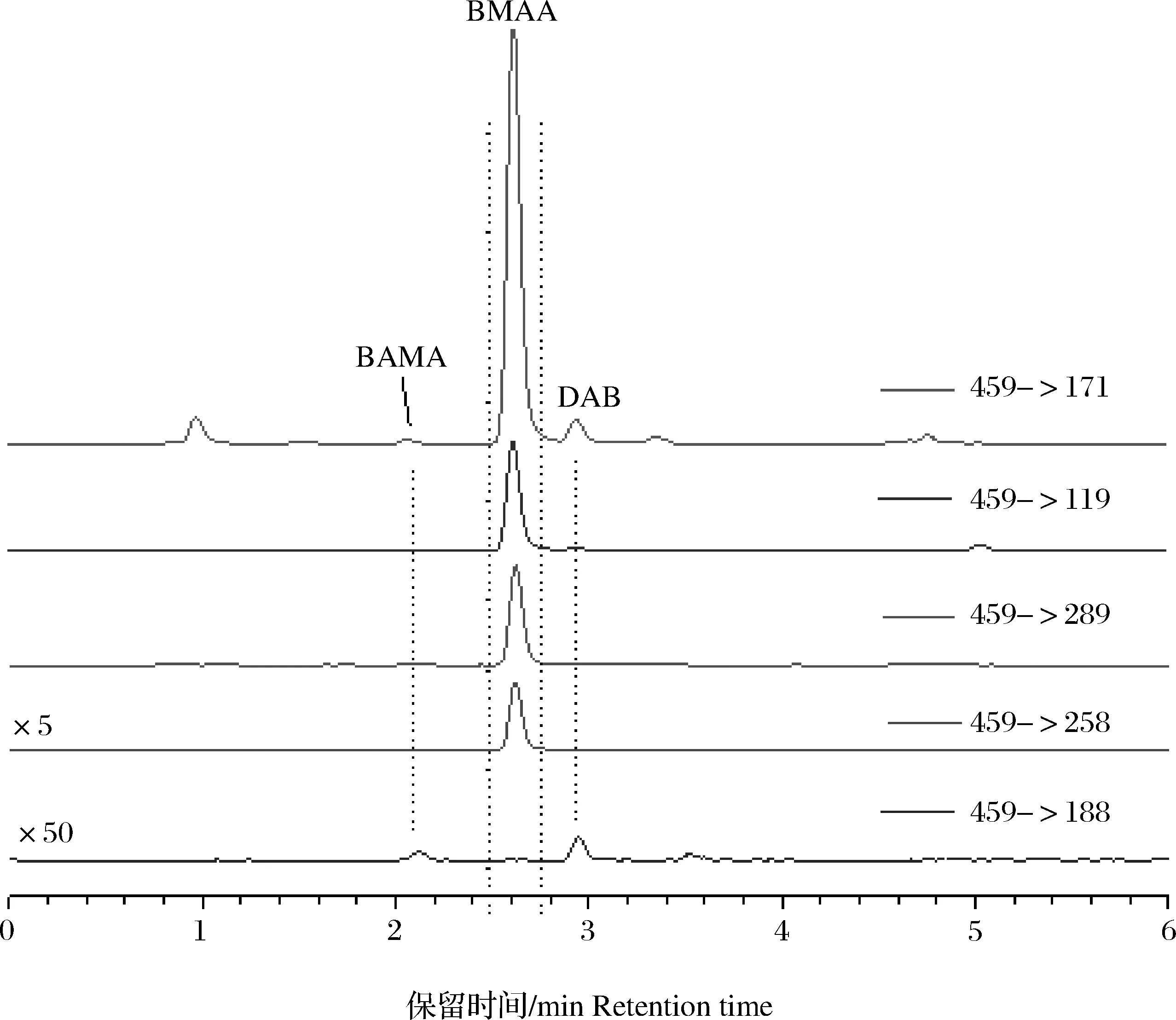
图3 AQC衍生法(Agilent 6430质谱)分析扁玉螺样品中BMAA及同系物的色谱图Y轴范围是0~3 000。Fig.3 Chromatograms of BMAA and its isomers in the gastropod samples using AQC-derivation method by Agilent 6430 mass spectrometerThe range of Y-axis is 0~3 000.
HILIC直接分析法和AQC-衍生法定量分析扁玉螺样品中BMAA和DAB的结果见表3。从表中可以看出,2种方法在扁玉螺样品中均检出了游离态BMAA和总溶解态BMAA及痕量的DAB毒素,但均未检出沉淀结合态BMAA和DAB以及任何形态的AEG。比较2种方法的定量分析结果,总体而言直接分析法获得的BMAA毒素的浓度结果普遍高于AQC-柱前衍生方法,2种方法分析游离态BMAA和总溶解态BMAA的定量结果的比值分别为0.83~2.57 和1.82~9.75,由此来看盐酸水解后的样品基质使得2种方法的定量结果偏差更大,这可能是因为可溶性的多肽在水解后释放出大量的氨基酸分子干扰BMAA与AQC分子的衍生效率,导致BMAA的回收率降低。比较2种不同的提取比例对定量分析结果的影响,使用高的溶剂提取比例(50∶1)时直接分析法获得的游离态BMAA和总溶解态BMAA的定量结果分别增加了(0.730±0.035)倍和(0.800±0.082)倍,AQC-衍生法在提高溶剂提取比例后2种形态BMAA的定量结果分别增加了(2.45±0.45)倍和(4.21±3.26)倍。由此来看,增加毒素提取溶剂的比例可以降低样品基质的抑制效应,使得毒素的定量结果升高,且对AQC-衍生法的定量结果影响更大。比较总溶解态和游离态BMAA浓度发现,HILIC直接分析法获得的总溶解态BMAA浓度高出(5.71±1.72)倍,AQC-衍生法获得的总溶解态BMAA的浓度高出(1.53±0.83)倍。由此来看,扁玉螺样品中平均85%的总溶解态BMAA是以溶解结合态形式存在的,但AQC-衍生法测定样品中溶解结合态BMAA的浓度偏低,这应该与水解后样品中氨基酸分子的干扰有关。
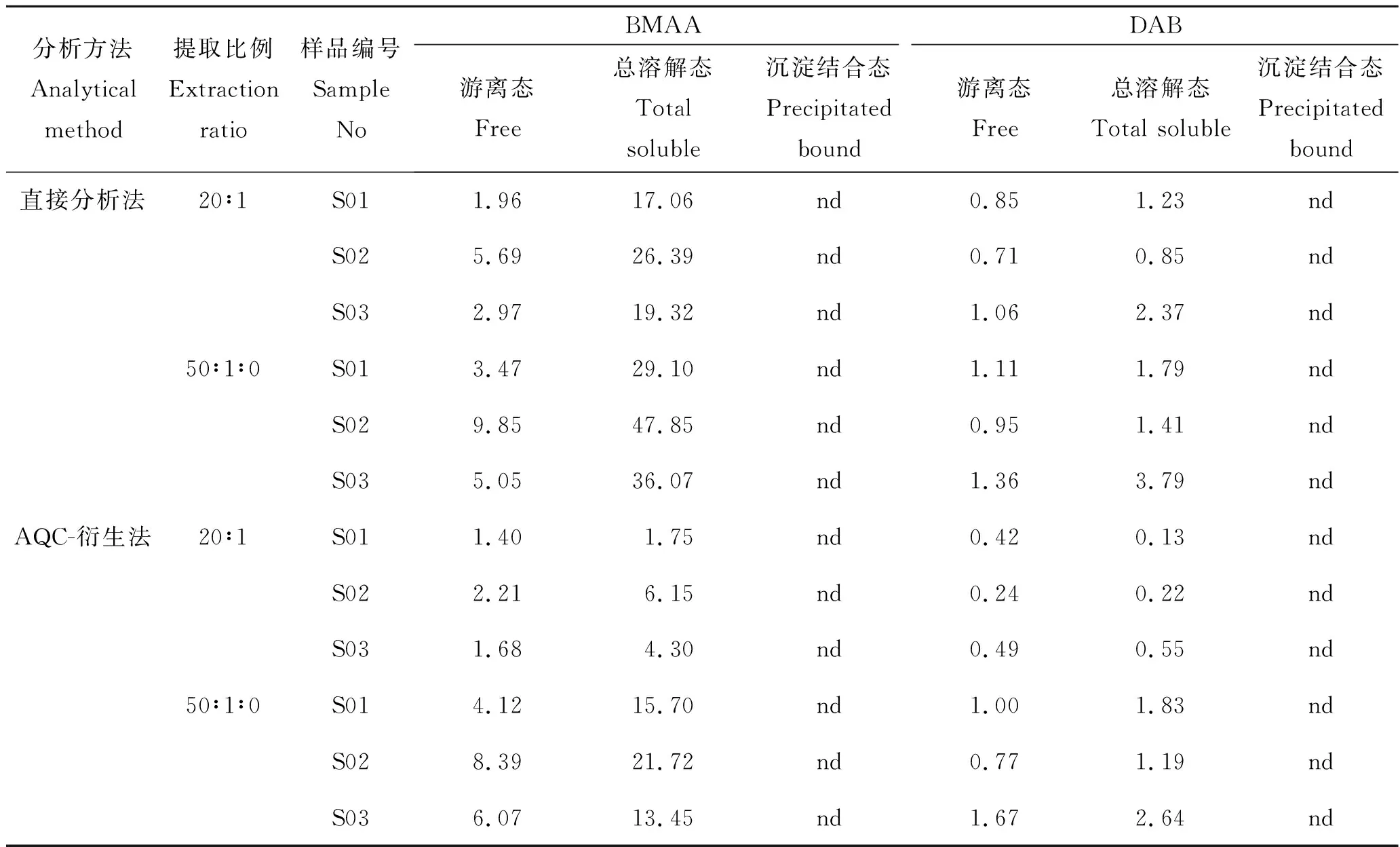
表3 扁玉螺样品中BMAA和DAB的分析结果(干重)Tab.3 Analytical results of BMAA and DAB in the gastropod samples (d.w.) μg·g-1
综上,研究结果证明,在分析扁玉螺样品中BMAA毒素时应充分搅碎贝肉组织,取少量的贝肉使用较高的溶剂提取比例制备样品,采用HILIC直接分析法定量分析样品中的总溶解态BMAA毒素,即可反映贝类样品中BMAA毒素的总量。
2.2 其他贝类样品中BMAA及其同系物的分析
为了更好地分辨贝类生物样品中的BMAA 和BAMA,本研究使用SeQuant ZIC HILIC色谱柱建立的LC-MS/MS方法对采自青岛近海的脉红螺、栉江珧和菲律宾蛤仔3种贝类样品中BMAA及其同分异构体化合物进行了分析(图4)。从图4可知,标准毒素中的BMAA、DAB和AEG在21~24 min之间可以实现基线分离,3种贝类样品在保留时间21 min前后均出现BMAA毒素的特征变迁离子119->88,但变迁离子119->56和119 ->102的色谱峰均出现分叉现象,说明存在其他化合物与BMAA毒素同时被洗脱下来的可能,且该化合物对BMAA的变迁离子119->88和119->44未见干扰,同时该化合物也无变迁离子119->101的色谱峰。
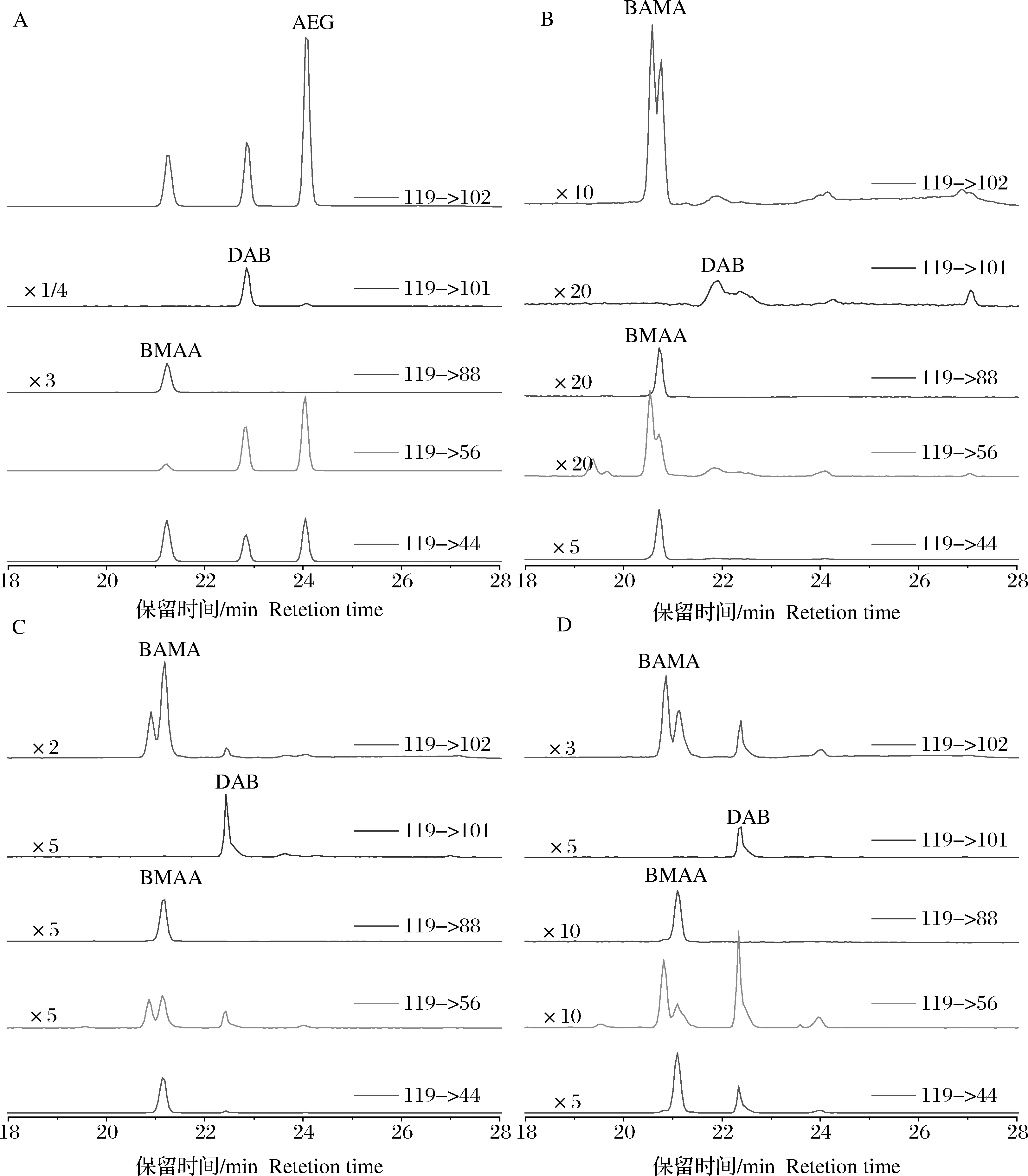
图4 LC-MS/MS(SeQuant ZIC HILIC色谱柱)分析(A)0.25 μg·mL-1混合标准、(B)脉红螺、(C)栉江珧和(D)菲律宾蛤仔提取液中BMAA及其同系物的色谱图Fig.4 LC-MS/MS chromatograms of BMAA and its isomers in (A) mixed standards (0.25 μg·mL-1), (B) Rapana venosa, (C) Atrina pectinata, and (D) Ruditapes philippinarum using an SeQuant ZIC HILIC column
据Foss等[24]报道,SeQuant ZIC HILIC色谱柱洗脱BMAA及其同分异构体时,BAMA比BMAA的洗脱体积略小,且BAMA仅对变迁离子119->102和119->56造成干扰[34],故本研究采用变迁离子119->44和119->88的峰面积的比值辅助确证BMAA(表4)。脉红螺、栉江珧和菲律宾蛤仔样品中二者的色谱峰峰面积的比值分别为2.30、2.25和2.29,与标准毒素的响应比值2.31基本吻合,因此推测3种生物样品中均含有一定量的BAMA与BMAA。另外,采用亲水交互作用色谱柱分析生物样品中的BMAA时,BMAA保留时间通常会受样品基质干扰而发生漂移[35]。比较标准毒素和样品中BMAA的保留时间发现,样品中BMAA毒素在SeQuant ZIC HILIC色谱柱上的洗脱体积略有减小,保留时间稍稍提前(图4)。因此,在甄别BMAA色谱峰的过程中应同时参考保留时间、特征变迁离子及变迁离子信号强度之间的比值。在应用LC-MS/MS直接分析生物样品中BMAA毒素的过程中,应首先注意BAMA和BMAA之间的区别,选择不受干扰的特征变迁离子119->88的峰面积进行毒素浓度的计算。

表4 LC-MS/MS(SeQuant ZIC HILIC色谱柱)分析不同贝类样品中BMAA的质谱响应强度的比值和保留时间的漂移Tab.4 Ratios of signal intensity and the retention time drift of BMAA in different mollusk samples using an SeQuant ZIC HILIC column
为了确认3个贝类样品中干扰BMAA检测的化合物,采用AQC-衍生方法对其进行了验证分析(图5)。从图5可以看出,3个样品均在保留时间2.4 min位置出现了一个清晰的色谱峰,除变迁离子459->258之外,这个化合物具有459->171、459->289和459->119 3个变迁离子色谱峰,且这3个峰的相对响应强度与BMAA毒素相似,只是459->289和459->119这2个变迁离子的峰面积更相近。比较标准毒素和3种不同贝类样品中不同变迁离子的相对响应强度(变迁离子的峰面积占4个变迁离子峰面积之和的百分比)发现(表5),BMAA标准毒素的变迁离子459->171色谱峰的峰面积最大,占总面积的62.03%,459->289和459->119次之,特征变迁离子459->258的色谱峰最小,占总面积的2.49%;与标准毒素相比,脉红螺、栉江珧和菲律宾蛤仔3种贝类样品中的4个变迁离子的色谱峰的峰面积所占比例基本吻合,保留时间也均为3.10 min,可以确认贝类样品含有BMAA。与HILIC色谱柱上BMAA毒素的保留时间相比,BMAA衍生物在C18色谱柱上的洗脱体积更稳定,未见样品基质效应的明显影响,这与亲水交互作用色谱柱分离毒素原理的缺陷有关。据报道,采用C18色谱柱和4组不同的流动相组成,分析软体动物中BMAA及其同分异构体,其中BAMA与BMAA保留时间的比值范围为0.76~0.79[36],而本研究中脉红螺、栉江珧和菲律宾蛤仔样品中BAMA与BMAA衍生物的保留时间比值均为0.77(2.4 min/3.1 min),进一步说明2.4 min洗脱出的化合物为BAMA的衍生物。因此,前面使用HILIC色谱柱直接分析3种贝类样品时BMAA色谱峰出现的分叉正是由于BAMA的共洗脱造成的。
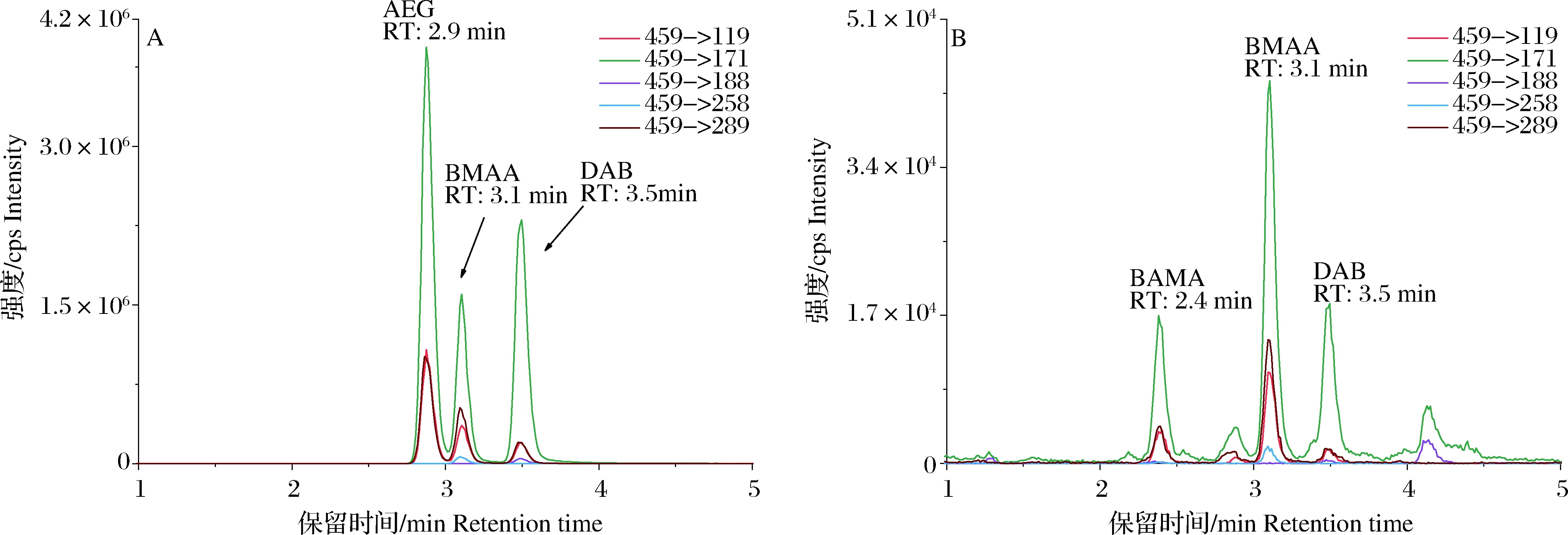
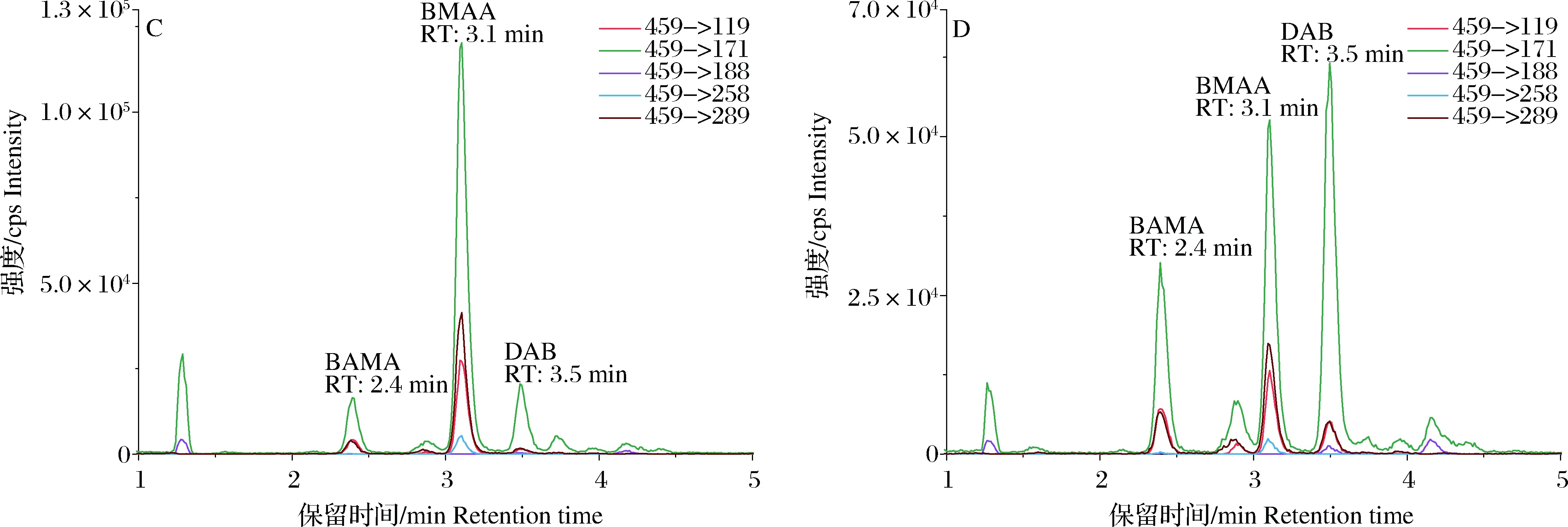
图5 AQC-衍生法分析(A)0.20 μg·mL-1混合标准、(B)脉红螺、(C)栉江珧和(D)菲律宾蛤仔样品中BMAA及其异构体化合物的色谱图(RT为保留时间)Fig.5 LC-MS/MS chromatograms of BMAA and its isomers using the AQC-derivation method in (A) mixed standards (0.20 μg·mL-1), (B) Rapana venosa, (C) Atrina pectinata, and (D) Ruditapes philippinarum (RT means retention time)
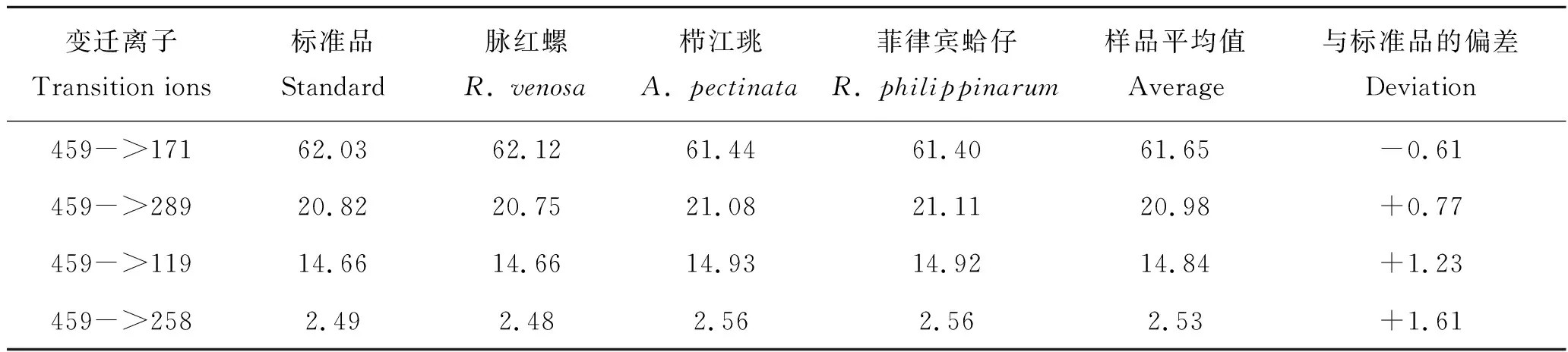
表5 AQC-衍生法分析不同贝类样品中BMAA变迁离子的色谱峰面积百分比Tab.5 Intensity percents of the transition ions of BMAA in different mollusk samples using AQC-derivation method%
本研究使用HILIC直接分析和AQC-柱前衍生2种方法在脉红螺、栉江珧和菲律宾蛤仔样品中均检出了BMAA和DAB毒素,其总溶解态毒素的含量如表6所示。从表中可以看出,栉江珧中BMAA的含量最高,其次是菲律宾蛤仔和脉红螺;HILIC直接分析法获得相比AQC-柱前衍生法的定量结果总体偏高,2种方法获得的定量结果的平均比值为(1.83±0.32),该比值低于2种方法分析扁玉螺样品中总溶解态BMAA结果的比值(1.82~9.75)。这可能是因为使用SPE纯化过程去除了贝类样品中的酸性氨基酸成分,降低了氨基酸分子对AQC-衍生法回收率的影响。另外,这3种贝类样品的溶剂提取比例是新鲜贝样的20∶1(10 mL/0.5 g湿重),如果按贝肉组织中含水率为80%计算,相当于冷冻干燥样品的溶剂提取比例100∶1,高于前文扁玉螺样品提取过程中使用的50∶1。因此,2种方法分析BMAA毒素定量结果偏差的缩小,也可能与使用了更高的溶剂提取比例有关。有关溶剂提取比例对不同贝类样品中基质效应的影响,有待今后系统研究。
前期研究表明AQC-柱前衍生法分析生物样品中BMAA时[36-38],BMAA及其同分异构体化合物的衍生物能够在C18色谱柱上实现基线分离,但AQC衍生方法的原理适用于生物样品中广泛存在的含有伯氨基和仲氨基的化合物,导致该方法对BMAA的特异性降低,易受生物样品基质中其他氨基酸分子的干扰。而HILIC-MS/MS直接分析BMAA的方法虽具有选择性高、灵敏度高的特点,但目标化合物在HILIC色谱柱上的洗脱体积易受样品基质的影响,且BAMA与BMAA难以实现基线分离,应注意采用多种标准判断BMAA的色谱峰。综合这两种方法分析BMAA及其异构体化合物的优劣,本研究建议使用HILIC色谱柱直接分析法对BMAA进行定性和定量分析,采用AQC-柱前衍生法辅助定性。
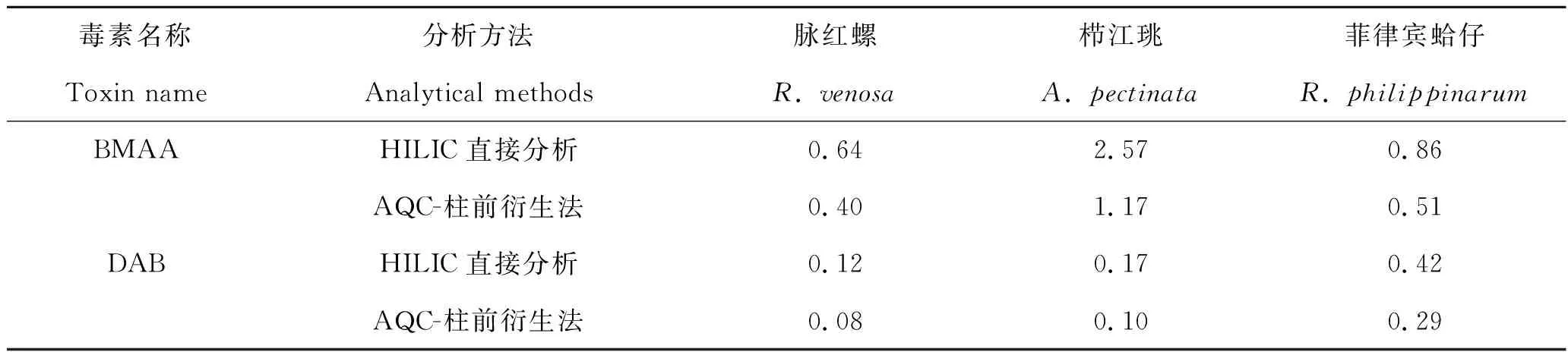
表6 脉红螺、栉江珧和菲律宾蛤仔体内BMAA和DAB毒素的含量(湿重)Tab.6 Contents of BMAA and DAB in Rapana venosa, Atrina pectinata, and Ruditapes philippinarum (wet weight) μg·g-1
3 结论
本研究基于目前流行的两种分析生物样品中BMAA及其同系物的LC-MS/MS方法,分别采用直接分析法和AQC-柱前衍生法分析了BMAA的典型携带生物扁玉螺及其他3种贝类样品,比较了2种方法分析贝类样品中BMAA及其同系物的优缺点。研究发现,贝类样品中的BAMA与BMAA毒素在2种亲水相互作用色谱柱上不能实现基线分离,但BMAA特征变迁离子119->88不受BAMA干扰,可采用变迁离子119->88色谱峰的峰面积进行样品的定量分析;AQC-柱前衍生法获得的BMAA及BAMA、DAB和AEG同系物的衍生物在C18色谱柱上可以实现基线分离,且目标化合物的保留时间不易受样品基质的干扰,但BMAA和DAB的定量结果普遍低于HILIC-MS/MS直接分析法的定量结果。分析贝类样品中BMAA时,可采用HILIC色谱柱直接分析法对BMAA进行定性和定量分析,采用AQC-柱前衍生法辅助定性。
