怀牛膝多糖的柱前衍生化-HPLC指纹图谱建立及单糖成分含量测定
2021-03-02王小燕郭常润常军民陈丽
王小燕 郭常润 常军民 陈丽

摘 要 目的:建立懷牛膝多糖的柱前衍生化-高效液相色谱(HPLC)指纹图谱,测定其主要单糖成分的含量,为怀牛膝饮片的质量评价提供参考。方法:以10批不同生产厂家的怀牛膝饮片为样品,采用水提醇沉法提取其多糖后,以三氟乙酸进行水解,再采用1-苯基-3-甲基-5-吡唑啉酮衍生化处理后,进行HPLC分析。色谱柱为Hanbon Hedera C18,柱温为30 ℃,流速为1.2 mL/min,流动相为乙腈-0.02 mol/L乙酸铵溶液(梯度洗脱),检测波长为250 nm,进样量为20 μL。采用《中药色谱指纹图谱相似度评价系统》(2012A版)建立10批怀牛膝多糖的HPLC指纹图谱并进行相似度评价,通过与对照品比对进行共有峰指认,并采用SPSS 25.0软件进行聚类分析。采用HPLC法对指认的单糖成分进行含量测定。结果:10批怀牛膝多糖样品的相似度均大于0.95;共标定了9个共有峰,并指认了其中5个共有峰,分别为无水葡萄糖(峰1)、甘露糖(峰2)、鼠李糖(峰4)、半乳糖醛酸(峰5)、阿拉伯糖(峰7)。聚类分析结果显示,编号为S1的样品单独为一类,编号为S2、S5、S8和S9的样品聚为一类,编号为S3、S4、S6、S7和S10的样品聚为一类。含量测定结果显示,10批样品中无水葡萄糖的含量为6.17~17.55 mg/g、甘露糖的含量为3.31~7.66 mg/g、鼠李糖的含量为38.80~73.97 mg/g、半乳糖醛酸的含量为2.49~8.95 mg/g、阿拉伯糖的含量为11.30~28.58 mg/g。结论:本研究建立的柱前衍生化-HPLC指纹图谱及单糖组分的含量测定方法可为怀牛膝饮片质量评价提供参考。
关键词 怀牛膝多糖;单糖;柱前衍生化;高效液相色谱法;指纹图谱;含量测定
中图分类号 R284 文献标志码 A 文章编号 1001-0408(2021)03-0294-07
DOI 10.6039/j.issn.1001-0408.2021.03.08
ABSTRACT OBJECTIVE: To establish pre-column derivatization-HPLC fingerprint of polysaccharide from Achyranthes bidentata,and to determine the contents of monosaccharide components,so as to provide reference for quality evaluation of A. bidentata decoction pieces. METHODS: Taking 10 batches of A. bidentata decoction pieces from different manufacturers as samples, the polysaccharides were extracted by water extraction and alcohol precipitation, hydrolyzed by trifluoroacetic acid, and then derivatized by 1-phenyl-3-methyl-5-pyrazolone for HPLC analysis. The determination was performed on Hanbon Hedera C18 column with column temperature of 30 ℃ at the flow rate of 1.2 mL/min. The mobile phase consisted of acetonitrile-0.02 mol/L ammonium acetate solution (gradient elution). The detection wavelength was set at 250 nm, and sample size was 20 μL. HPLC fingerprint was established and similarity evaluation was performed for 10 batches of A. bidentata polysaccharide by using TCM Chromatogramic Fingerprint Similarity Evaluation System (2012A edition). The common peak was identified by comparing with the reference substance,and cluster analysis was performed by using SPSS 25.0 software. The contents of identified monosaccharides were determined by HPLC. RESULTS: The similarity of 10 batches of A. bidentata polysaccharide were all higher than 0.95. Nine common peaks were fixed and a total of 5 common peaks were identified, namely anhydrous glucose (peak 1), mannose (peak 2), rhamnose (peak 4), galacturonic acid (peak 5) and arabinose (peak 7). Results of cluster analysis showed that S1 sample was classified into one category; S2, S5, S8 and S9 samples were clustered into one category; S3, S4, S6, S7 and S10 samples were clustered into one category. Results of content determination showed that the contents of anhydrous glucose in 10 batches of samples ranged from 6.17 to 17.55 mg/g; those of mannose ranged from 3.31 to 7.66 mg/g; those of rhamnose ranged from 38.80 to 73.97 mg/g; those of galacturonic acid ranged from 2.49 to 8.95 mg/g; those of arabinose ranged from 11.30 to 28.58 mg/g. CONCLUSIONS: Established pre-column derivatization HPLC fingerprints and content determination method can provide reference for quality evaluation of A. bidentata decoction pieces.
KEYWORDS Achyranthes bidentata polysaccharide; Monosaccharide; Pre-column derivatization; HPLC; Fingerprint; Contnet determination
怀牛膝Achyranthes bidentata BI.为常用的大宗中药材之一,最早出自《神农本草经》,以根入药,主要含有甾酮类、皂苷类、多糖类等化合物[1-2]。据文献报道,怀牛膝多糖类组分的毒性低、水溶性好,具有抑制肿瘤细胞转移、增强免疫力、保护肝脏等多种药理活性[3]。而多糖通常由各种相对固定比例的单糖组成[4-5],因此分析多糖中单糖的组成对于研究多糖质量标准和探索多糖结构、性质及构效关系等信息具有重要意义[6]。
关于多糖中单糖组分的分析方法,通常是先用三氟乙酸将多糖水解成单糖,再利用发光试剂衍生后通过高效液相色谱(HPLC)法进行分离和检测[7]。而指纹图谱作为一种可体现中药化学成分整体特征的质量评价方法,对药材的全面控制具有重要作用[8]。若将生物大分子多糖经衍生化后分析其单糖组成,再结合指纹图谱技术,可以更加全面、整体地评价药材质量[9-10]。笔者查阅文献后发现,目前有关怀牛膝多糖的研究主要集中于总多糖的提取工艺及生物活性[11-14],而对于其中单糖组成及含量分析的研究尚未见相关报道。因此,本研究拟建立怀牛膝多糖的1-苯基-3-甲基-5-吡唑啉酮(PMP)柱前衍生化-高效液相色谱(HPLC)指纹图谱,分析其单糖组成并测定单糖组分的含量,为怀牛膝饮片的质量评价提供参考。
1 材料
1.1 主要仪器
本研究所用主要仪器有1260型HPLC仪(包括G1312C型双元泵、G1329B型检测器、G1316A型柱温箱、G1315D型进样器,美国Agilent公司)、BSA124S型万分之一电子天平(北京赛多利斯仪器系统有限公司)、TGL-16型台式高速冷冻离心机(长沙湘仪离心机仪器有限公司)、BDHH-6型数显恒温水浴锅[贝蒂(上海)仪器有限公司]、XW-80A微型旋涡混合仪(上海沪西分析仪器厂有限公司)、BHX型电热恒温鼓风干燥箱(南京科尔仪器设备有限公司)、PHS-25型数显pH计(上海仪电科学仪器股份有限公司)等。
1.2 主要药品与试剂
本研究所用的药品与试剂主要有无水葡萄糖对照品、D-半乳糖醛酸对照品、鼠李糖对照品、D-甘露糖对照品、L(+)阿拉伯糖对照品(中国食品药品检定研究院,批号分别为110833-201707、140648-201804、111646- 201702、111508-201605、140651-201805,纯度均不低于98%),D-葡萄糖醛酸(上海源叶生物科技有限公司,批号1506-200202,纯度≥98%);乙腈为色谱纯,乙酸铵、氢氧化钠、三氟乙酸、盐酸、磷酸二氢钾、PMP、甲醇、氯仿等均为分析纯,水为娃哈哈纯净水。
10批怀牛膝饮片购自南京市各大药房(来源信息见表1),经中国药科大学中药学院杨中林教授鉴定为怀牛膝A. bidentata BI.的干燥根。样品保存于中国药科大学中药学院中药炮制实验室。
2 方法与结果
2.1 怀牛膝多糖的制备
称取各批次怀牛膝饮片约10 g,将其粉碎过60目筛,并以80%乙醇为溶剂[料液比1 ∶ 12(g/mL)]在80 ℃水浴中回流提取2次,每次2 h;弃去滤液,滤渣挥干溶剂后称质量,然后继续以水为溶剂[料液比1 ∶ 15(g/mL)]在98 ℃水浴中回流提取3次,每次2 h;合并3次水提取液,抽滤,合并滤液后浓缩至50 mL,冷却。缓慢向浓缩液中加入无水乙醇至乙醇终体积分数为70%,室温静置24 h,抽滤后得沉淀物1;然后将滤液浓缩至50 mL,再次加入无水乙醇至乙醇终体积分数为80%,室温下静置24 h,抽滤得沉淀物2。合并沉淀物1和沉淀物2,用丙酮和无水乙醇交替洗涤,直至洗液变为无色为止,然后干燥除去残留的洗涤试剂后,得到的沉淀物即为怀牛膝多糖。以D-无水葡萄糖为标准品,采用苯酚-硫酸法[15]测得10批怀牛膝饮片(编号S1~S10)中多糖的得率分别为18.10%、18.30%、17.56%、10.74%、17.52%、16.08%、15.88%、13.2%、14.8%、16.52%。
2.2 溶液的制备
2.2.1 单糖对照品溶液 称取D-无水葡萄糖对照品6.87 mg、D-甘露糖对照品5.51 mg、鼠李糖对照品5.70 mg、D-半乳糖醛酸对照品4.66 mg、L(+)阿拉伯糖对照品4.35 mg,置于不同1 mL量瓶中,分別用水溶解并稀释至刻度,涡旋混匀,即得各单糖的单一对照品溶液。
2.2.2 怀牛膝多糖水解液 精密称取100 mg怀牛膝多糖至10 mL离心管中,加入2 mol/L三氟乙酸溶液4 mL使溶解,密封后,在110 ℃烘箱中水解4 h;取出,冷却至常温,并以3 mol/L NaOH溶液调节水解液的pH至6.5,即得。
2.2.3 阴性对照溶液 除不加多糖样品外,其余按“2.2.2”项下方法操作,制得阴性对照溶液。
2.3 衍生化处理及各衍生化溶液制备
精密吸取“2.2.1”项下各单糖对照品溶液、“2.2.2”项下多糖水解液和“2.2.3”项下阴性对照溶液各0.3 mL,置于不同5 mL离心管中,分别加入0.5 mol/L PMP甲醇溶液300 ?L和0.3 mol/L NaOH溶液300 ?L,涡旋混合,在70 ℃水浴中衍生30 min;待衍生完毕放冷后,加入0.3 mol/L HCl溶液300 ?L终止反应。在离心管中加入1 mL氯仿溶液进行萃取,弃去氯仿层,取上层溶液;再如上重复操作3次,合并上层溶液后以0.35 ?m滤膜滤过,即得到经衍生化处理后的各溶液。分别吸取经衍生化处理后的各单糖对照品溶液 0.2 mL,充分混合后,经0.35 ?m滤膜过滤,收集滤液,即得经衍生化处理后的混合对照品溶液。
2.4 色谱条件与系统适用性试验
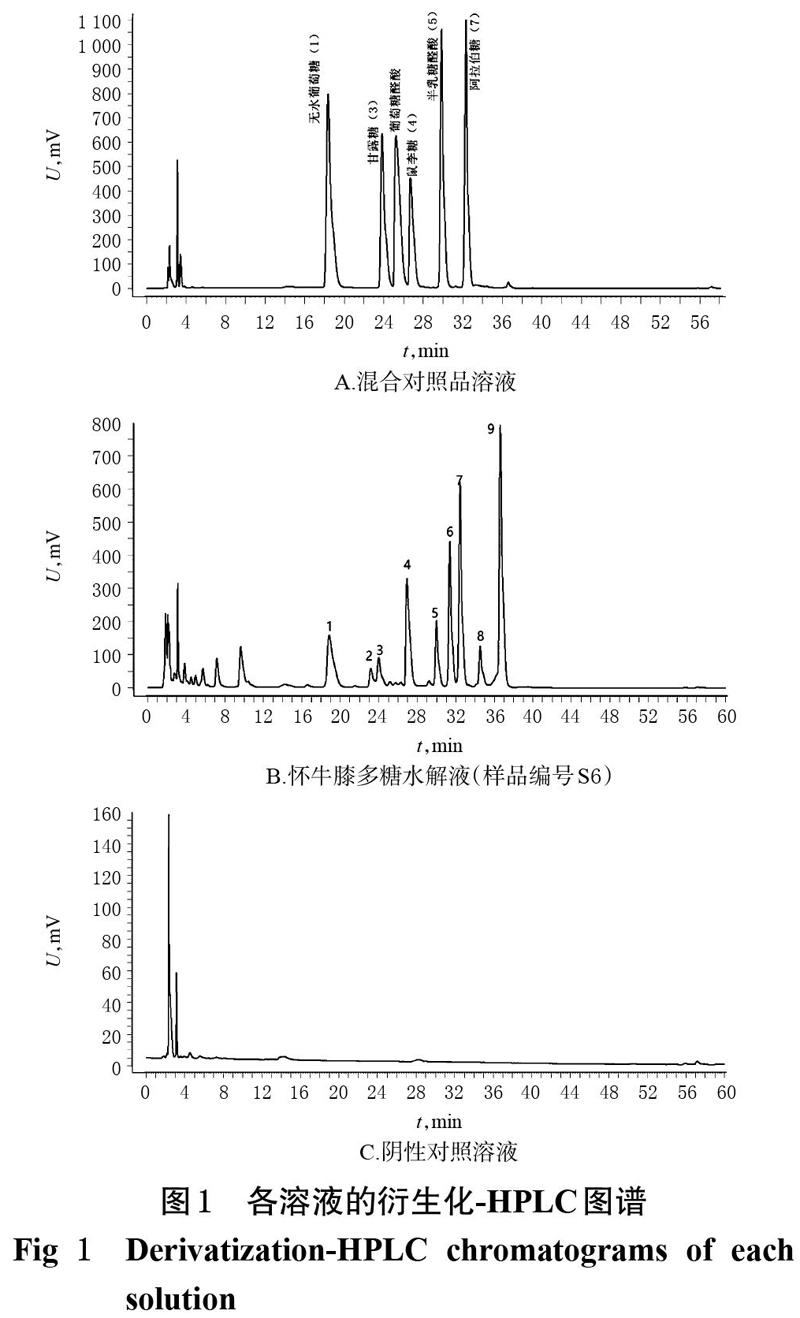
色谱柱为Hanbon Hedera C18(250 mm×4.6 mm,5 μm);以乙腈(A)-0.02 mol/L乙酸铵溶液(B)为流动相进行梯度洗脱(0~15 min,19%A;15~50 min,19%A→35%A;50~55 min,35%A→42%A;55~55.1 min,42%A→19%A;55.1~60 min,19%A);柱温为30 ℃;流速为1.2 mL/min;检测波长为250 nm;进样量为20 μL。取按“2.3”方法衍生化处理的怀牛膝多糖水解液(样品编号S6)、阴性对照溶液和混合对照品溶液,分别在此色谱条件下进样测定,记录色谱图。结果,在此色譜条件下,各成分峰均能达到良好分离(分离度均大于1.5),理论板数以甘露糖计大于6 000,且阴性对照溶液对样品峰的测定不产生干扰。各溶液的衍生化-HPLC图谱见图1。
2.5 指纹图谱的建立与分析
2.5.1 精密度试验 取100 mg怀牛膝多糖(样品编号S6),按“2.2.2”“2.3”项下方法水解并衍生化处理后,再按“2.4”项下色谱条件进样分析,连续测定6次,记录峰面积。以鼠李糖(峰4,该峰分离度良好且稳定、峰位相对居中)为参照峰,计算得各共有峰相对保留时间和相对峰面积的RSD均小于3.0%(n=6),表明该方法精度良好。
2.5.2 重复性试验 取100 mg怀牛膝多糖(样品编号S6),共6份,分别按“2.2.2”“2.3”项下方法水解并衍生化处理后,再按“2.4”项下色谱条件进样测定,记录峰面积。以鼠李糖(峰4)为参照峰,计算得各共有峰相对保留时间和相对峰面积的RSD均小于3.0%(n=6),表明该方法重复性较好。
2.5.3 稳定性试验 取100 mg怀牛膝多糖(样品编号S6),分别按“2.2.2”“2.3”项下方法水解并衍生化处理后,分别于室温放置0、2、4、8、12、24 h 时,按“2.4”项下色谱条件进样测定,记录峰面积。以鼠李糖(峰4)为参照峰,计算得各共有峰相对保留时间和相对峰面积的RSD均小于3.0%(n=6),表明该溶液在室温下放置24 h内稳定。
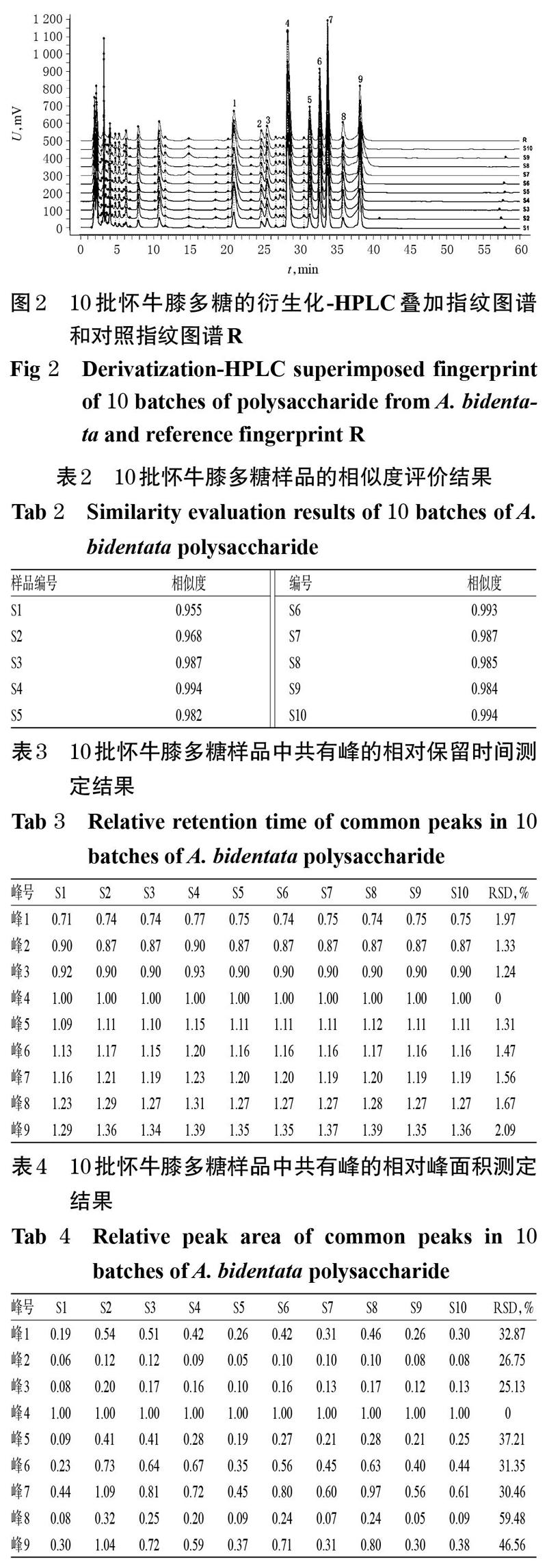
2.5.4 指纹图谱的建立及样品相似度评价 分别称取10批怀牛膝多糖适量(样品编号S1~S10),按“2.2.2”“2.3”项下方法水解并衍生化处理后,再按“2.4”项下色谱条件进样测定,记录色谱图。利用《中药指纹图谱相似度系统评价软件》(2012A版)对S1~S10多糖样品的HPLC指纹图谱进行分析,选择编号为S1的多糖样品的图谱作为参照图谱(各峰稳定且分离度较好),将时间宽度设置为0.1 min、间距设置为50,通过多点校准法生成10批多糖样品的叠加指纹图谱,并使用中值法生成对照指纹图谱(R)。以对照指纹图谱R为参照,对10批样品的相似度进行整体评价。结果,10批样品的相似度为0.955~0.994;共得到了9个共有峰,以峰4为参照峰计算得各共有峰相对保留时间的RSD为0~2.09%(n=10)、相对峰面积的RSD为0~59.48%(n=10)。该结果提示,10批怀牛膝多糖样品的共有成分相似,但含量存在差异。10批怀牛膝多糖的衍生化-HPLC叠加指纹图谱和对照指纹图谱见图2,相似度评价结果见表2,各共有峰的相对保留时间、相对峰面积测定结果分别见表3、表4。
2.5.5 色谱峰的指认 通过将对指纹图谱R(见图3)与混合对照溶液衍生化-HPLC图谱(见图1A)的色谱峰进行比对,指认了其中5个共有色谱峰,分别为无水葡萄糖(峰1)、甘露糖(峰2)、鼠李糖(峰4)、半乳糖醛酸(峰5)和阿拉伯糖(峰7)。
2.6 聚类分析
将10批怀牛膝多糖衍生化-HPLC图谱中9个共有峰的峰面积分别导入SPSS 25.0软件中,采用组间连接方法,以平方欧氏距离作为测量间隔进行系统聚类分析。结果显示,当平方欧氏距离为5时,10批怀牛膝多糖被聚为3类:编号为S1的样品单独为一类,相似度为0.955;编号为S2、S5、S8和S9的样品聚为一类,相似度在0.968~0.985之间;编号为S3、S4、S6、S7和S10的样品聚为一类,相似度在0.987~0.994之间。10批怀牛膝多糖样品的聚类分析树状图见图4。
2.7 单糖成分的含量测定
2.7.1 线性关系考察 取“2.2.1”项下各单糖对照品溶液各0.3 mL,分别按“2.3”项下方法进行衍生化处理后,分别取0.2 mL各单糖衍生液于同一10 mL离心管中,充分混匀,即得衍生化处理后的混合对照品溶液。分别吸取该混合对照品溶液0.5、0.45、0.35、0.25、0.15、0.05、0.008 mL于不同的1 mL量瓶中,加甲醇定容,即得系列不同质量浓度的溶液,分别按“2.4”项下色谱条件下进样测定,记录峰面积。以各待测成分的峰面积(y)为纵坐标、质量浓度(x,mg/mL)为横坐标绘制标准曲线。结果,5种单糖在各自质量浓度范围内的线性关系均良好(r均大于0.999 0),结果见表5。
2.7.2 精密度试验 取“2.2.1” 项下的单糖对照品溶液各0.3 mL,按“2.3”项下方法衍生化处理后,分别取各单糖衍生液0.2 mL于同一10 mL离心管中,充分混匀,制得混合对照品溶液,然后按 “2.4”色谱条件进样测定,连续测定6次,记录峰面积。结果,无水葡萄糖、甘露糖、鼠李糖、半乳糖醛酸和阿拉伯糖峰面积的RSD分别为1.59%、2.78%、2.35%、2.44%、1.98%(n=6),表明仪器精密度良好。
2.7.3 稳定性试验 取100 mg怀牛膝多糖(样品编号S6),分别按“2.2.2”“2.3”项下方法水解并衍生化处理后,于室温放置0、2、4、8、12、24 h时,再按“2.4”项下色谱条件进样测定,记录峰面积。结果,无水葡萄糖、甘露糖、鼠李糖、半乳糖醛酸和阿拉伯糖峰面积的RSD分别为2.35%、2.91%、1.37%、2.79%、1.39%(n=6),表明该溶液在室温下放置24 h内稳定性良好。
2.7.4 重复性试验 取100 mg怀牛膝多糖(样品编号S6),共6份,分别按“2.2.2”“2.3”项下方法水解并衍生化处理后,再按“2.4”项下色谱条件进样测定,记录峰面积,并根据标准曲线计算各待测成分的含量。结果,无水葡萄糖、甘露糖、鼠李糖、半乳糖醛酸和阿拉伯糖含量的RSD分别为2.17%、2.75%、1.94%、2.13%、2.86%(n=6),表明该方法的重复性良好。
2.7.5 加样回收率试验 取100 mg已知含量的怀牛膝多糖(样品编号S6),共6份,置于不同10 mL离心管中,分别精密加入与已知含量相近的相应对照品溶液,按“2.2.2”“2.3”项下方法水解并衍生化处理后,再按“2.4”项下色谱条件進样测定,记录峰面积,根据标准曲线计算出各待测成分的含量,并计算其加样回收率。结果,5种单糖的平均加样回收率为96.52%~99.15%,RSD均小于3.0%(n=6),表明该方法准确度较好,详见表6。
2.7.6 样品含量测定 分别取10批怀牛膝多糖,各约100 mg,精密称定,分别按“2.2.2”“2.3”项下方法水解并衍生化处理后,再按“2.4”项下色谱条件进样测定,记录峰面积,并根据标准曲线计算5种单糖的含量。每批样品平行测定3份,取平均值。结果,10批怀牛膝多糖样品中无水葡萄糖的含量为6.17~17.55 mg/g、甘露糖的含量为3.31~7.66 mg/g、鼠李糖的含量为38.80~73.97 mg/g、半乳糖醛酸的含量为2.49~8.95 mg/g、阿拉伯糖的含量为11.30~28.58 mg/g,表明不同批次怀牛膝多糖的单糖含量存在一定差异,详见表7。
3 讨论
3.1 色谱条件的优化
在单糖成分的分析过程中,笔者先考察了在230、240、250、260 nm这4个检测波长下的检测效果,发现在250 nm波长下所得色谱图的峰形尖锐,检测效果较在其他3个波长下更好,因此本研究选择250 nm为检测波长。此外,笔者分别考察了10、20 μL进样量对峰形的影响,发现进样量为20 μL时的色谱图峰形更好,故本研究以20 μL为进样量。笔者进一步考察了甲醇-水、乙腈-水、乙腈-0.1 mol/L磷酸盐溶液、乙腈-0.01 mol/L乙酸铵溶液、乙腈-0.02 mol/L乙酸铵溶液等流动相体系对检测效果的影响,发现水相为磷酸缓冲盐溶液时所需浓度较大,容易发生色谱柱堵塞,不利于仪器的使用及保护,因此采用乙酸铵溶液作为流动相的水相,并进一步确定0.02 mol/L为水相乙酸铵溶液的浓度。
3.2 混合对照品溶液制备方法的选择
本研究前期考察了2种方法制备的混合对照品溶液的衍生化效果:第一种是先将各单糖对照品溶液混合,制成混合对照品溶液后,再进行PMP衍生化处理;第二种是先将各单糖对照品溶液分别进行PMP衍生化处理制成单糖对照品衍生液后再进行混合。结果发现,采用第一种方法制备的混合对照品衍生液的HPLC色谱图峰形不对称,且部分峰无法分离,笔者推测这可能是混合后的单糖溶液在加热衍生过程中各单糖间又发生了反应。而本研究最终采用的第二种方法是先制备各单糖衍生液后再混合,可避免受热后各单糖间发生反应,故进样后所得色谱图中各成分的峰形对称且分离度良好。
3.3 多糖样品的预处理及指纹图谱的分析
指纹图谱分析目前主要用于中药的小分子成分,对分子量较大、结构复杂、无紫外吸收的多糖类物质不适用[16]。因此,为了提高多糖在HPLC检测中的灵敏度,多将多糖水解后再以发光试剂衍生化处理后,再使用HPLC法进行分离和检测[17-21]。本研究采用PMP为衍生试剂的原理是PMP在碱性条件下可以与单糖定量缩合成PMP-单糖衍生物,该物质相对稳定,在250 nm波长处具有强光吸收[22-25]。通过分析化合物中多糖的主要单糖组成和相对含量,可以识别不同来源多糖;通过测定多糖中主要单糖成分的绝对含量,可整体上评估不同批次药材的质量[26-27]。
本研究建立了10批不同厂家怀牛膝多糖的衍生化-HPLC指纹特征图谱,标定了9个共同峰,并指认出了5个特征峰,初步分析出怀牛膝多糖由无水葡萄糖、甘露糖、鼠李糖、半乳糖醛酸、阿拉伯糖等5种单糖构成,由此进一步明确了牛膝多糖中单糖的分布规律,为其多糖结构分析和质量控制提供了有力依据。但是也可能还存在部分未被识别的单糖,且仅从单糖组成的角度分析多糖仍具有一定的局限性,故后期笔者将从多糖的相对分子量、溶解性及构效关系方面对其进一步研究。
综上所述,本研究建立了怀牛膝多糖的PMP柱前衍生化-HPLC指纹图谱及主要单糖成分的含量测定方法,该方法精密度高、准确性好且分离效果较理想,可为怀牛膝饮片的进一步质量控制提供参考。
参考文献
[ 1 ] 胡婷婷,张振凌.中药牛膝化学成分、药理作用及储藏保管[J].中国老年学杂志,2016,36(13):3321-3322.
[ 2 ] 梁献葵,雷敬卫,龚海燕,等. HPLC测定怀牛膝及其芦头5种成分含量[J].中药材,2019,42(7):1578-1583.
[ 3 ] 李静.怀牛膝的研究进展[J].中国医药指南,2013,11(10):462-463.
[ 4 ] 李运,邱国玉,李晓春,等.基于主成分分析的前胡类药材HPLC指纹图谱研究[J].药物分析杂志,2019,39(7):1323-1329.
[ 5 ] 杜泽飞,陶爱恩,夏从龙,等.基于PMP-HPLC和化学计量学的黄精基原物种多糖差异分析[J].中国实验方剂学杂志,2019,25(15):25-29.
[ 6 ] 李薇,夏晴,孙成荣,等.柱前衍生(PMP)-HPLC法测定不同品种甘草多糖中单糖组成[J].辽宁中医药大学学报,2014,16(1):56-58.
[ 7 ] 周彦强,吴光斌,陈发河. PMP柱前衍生化HPLC法测定黄秋葵多糖的单糖组成[J].食品科学,2019,40(4):266- 271.
[ 8 ] 曹斯琼,吴文平,罗宇琴,等.王不留行炮制前后的UPLC指纹图谱比较及刺桐碱和王不留行黄酮苷的含量测定[J].中国药房,2020,31(19):2365-2370.
[ 9 ] 邹纯才,鄢海燕.我国中药色谱指纹图谱相似度评价方法30年(1988-2017年)研究进展与展望[J].中国中药杂志,2018,43(10):1969-1977.
[10] 汪海斌,石岩,李芳,等.中药黄芪指纹图谱的研究进展[J].中国药房,2017,28(33):4749-4752.
[11] 杨林松,钟慧,王璐瑶,等.怀牛膝多糖对RAW264.7细胞吞噬活性、表型和细胞因子表达的调节[J].常州大学学报(自然科学版),2020,32(4):55-62.
[12] 王媛媛,张梦凡,查丽丽,等.川牛膝和怀牛膝多糖提取工艺研究[J].长春师范大学学报,2016,35(10):77-81.
[13] 魏涛,何培新,封盛雪,等.怀牛膝多糖的超声波辅助提取工艺及其抗氧化性活性[J].食品与发酵工业,2011,37(2):191-194.
[14] 郭宁,郭婕.怀牛膝茎叶多糖提取及生物活性[J].周口师范学院学报,2017,34(2):120-122.
[15] 景永帅,马云凤,王非凡,等.丹参多糖的含量测定、结构表征及药理活性研究进展[J].中国药房,2020,31(16):2037-2042.
[16] 侯晓蓉,任立琴,裘飞君,等.黄芪生脉饮中多糖水解单糖指纹图谱的研究[J].浙江工业大学学报,2020,48(3):300-306.
[17] 李贝贝,卢燕.多糖色谱指纹图谱的研究方法进展[J].药学研究,2018,37(1):41-45.
[18] WANG XH,ZHAO J,LI DM,et al. Structural investigation of a uronic acid-containing polysaccharide from abalone by graded acid hydrolysis followed by PMP- HPLC-MSn and NMR analysis[J]. Carbohydr Res,2015.DOI:10.1016/j.carres.2014.10.010.
[19] FU DT,ONEILL RA. Monosaccharide composition analysis of oligosaccharides and glycoproteins by high-performance liquid chromatography[J]. Anal Bio Chem,1995,227(2):377-384.
[20] 陈燕文,李玉娟,宋梦璐,等. PMP柱前衍生化-HPLC法分析玛咖多糖的单糖组成[J].当代化工,2017,46(8):1513-1516.
[21] 姜彩霞,刘晓兰,田英华,等.高效液相色谱法测定亚麻纤维果胶单糖衍生条件的优化[J]. 毛纺科技,2014,42(10):1-4.
[22] 戴军,朱松,汤坚,等. PMP柱前衍生高效液相色谱法分析杜氏盐藻多糖的单糖组成[J].分析测试学报,2007,26(2):206-210.
[23] XUE S,WANG L,CHEN S,et al. Simultaneous analysis of saccharides between fresh and processed radix rehmanniae by HPLC and UHPLC-LTQ-Orbitrap-MS with multivariate statistical analysis[J]. Molecules,2018,23(3):541-560.
[24] JIANG WX,DI SX,HUA HM,et al. Structural characte- rization of polysaccharide from Cyclocarya paliurus leaves and its α-glucosidase inhibitory effect[J]. Chinese Traditional and Herbal Drugs,2017,48(8):1524-1528.
[25] 赖戈娜,贾文玉,罗思婉,等.猪苓多糖的PMP柱前衍生化-HPLC指纹图谱研究[J].中国药房,2020,31(7):788- 793.
[26] 张璐瑶,赵峡,陈欢欢.糖类化合物PMP衍生分析进展[J].分析测试学报,2016,35(3):367-372.
[27] 万强,吴学昊,范华均,等. HPLC衍生化法分析决明子多糖水解产物中单糖组分及其多糖组成特征的研究[J].分析测试学报,2014,33(11):1231-1236.
(收稿日期:2020-10-30 修回日期:2020-12-23)
(編辑:林 静)
