胸壁恶性蝾螈瘤一例手术治疗及文献复习
2021-03-02王海明方泽民丁志丹赵高峰
王海明,赵 凯,方泽民,丁志丹,赵高峰
郑州大学第一附属医院胸外科 郑州 450052
恶性蝾螈瘤(malignant triton tumor,MTT)是恶性外周神经鞘瘤(malignant peripheral nerve sheath tumors,MPNST)的一种罕见亚型,伴横纹肌肉瘤分化,含有神经组织和横纹肌组织2种成分,临床病程特别具有侵袭性。文献[1-4]报道,MPNST占所有软组织肉瘤的5%~10%;MTT占所有MPNST的5%[3-4]。因此,与MPNST相比,MTT的恶性程度更高,侵袭性更强,临床更为少见。有文献[5]报道,1973年至2018年,大约有162例MTT病例被报道,且大多数是病例报告。胸壁MTT更为少见,迄今为止,仅有8篇文献共9个胸壁MTT病例被报道[6-13],其中北京协和医院报道2例[6],台湾慈济医院报道1例[7],贵州医科大学报道1例[13]。作者现将郑州大学第一附属医院2019年12月8日收治的 1 例胸壁MTT患者的资料报道如下。
1 临床资料
1.1病例介绍患者,男,17岁,1 a前无意中发现右侧胸壁腋窝下出现一肿物,如枣大小,质软,边界清,活动度差,当时未在意,未到医院诊治。2个月前,肿块逐渐增大,遂到当地医院就诊。胸部CT示:右侧腋窝可见直径约7.1 cm的软组织肿块,边界清,周围组织受推移位。在当地医院行穿刺活检,病理结果显示:(右腋窝)梭形细胞肿瘤。为进一步诊治收入我院。查体:右颈部可触及肿大淋巴结,部分融合成团;右前外侧胸壁延伸至右腋下区域明显隆起,可触及一粗壮条索样肿块,大小约14 cm×8 cm×6 cm,无明显压痛,质韧,活动度差,右肩关节活动轻度受限。胸部增强CT示:右腋窝团块状软组织影,内密度欠均匀,可见不均匀轻度强化,边界欠清,最大截面大小约为93 mm×74 mm(图1A),邻近组织受压,临近骨质结构未见破坏,纵隔及左侧腋窝下未见明显肿大淋巴结影。颈部、腋窝、腹股沟淋巴结彩超及右胸壁彩超示:①右侧颈部Ⅲ、Ⅴ、Ⅵ区可见多个淋巴结回声,较大一个大小约16.0 mm×7.5 mm,部分融合,形状不规则,边界欠清,皮髓质分界不清;彩色多普勒血流显像(color Doppler flow imaging,CDFI)可见Ⅱ级血流信号。②左侧颈部Ⅴ区可见两个淋巴结回声,较大一个大小约8.8 mm×5.0 mm,边界欠清,皮髓质分界不清;CDFI可见点状血流信号。③右侧腋窝探及大小约94 mm×64 mm囊实性包块,边界清晰,回声不均匀,以等回声为主,内可见大小约26 mm×28 mm无回声区;CDFI显示等回声内可见Ⅰ级血流信号。④双侧腹股沟及左侧腋窝未见明显肿大淋巴结回声。颅脑MRI平扫示:右侧颞极前上方异常信号,考虑蛛网膜下腔增宽或蛛网膜囊肿。心脏、肝胆脾胰、双下肢深静脉彩超未见明显异常。实验室检验结果:①红细胞沉降率为38 mm/h。②血常规、凝血四项、传染病八项、肝肾功、血电解质等均无明显异常。
1.2治疗方法在征得患者及家属同意并签订手术知情同意书后,全麻下对患者实施了右胸壁肿瘤切除术。患者取平卧位,垫高右侧肩胸侧部,右上肢外展90°,充分暴露腋后线,自右锁骨下肿块上极至右腋窝肿块下极做一长约16 cm斜形切口,因肿块体积大、显露困难,于右侧胸大肌附着点断开部分胸大肌并掀开,术中见肿块大小约13 cm×6 cm×5 cm,呈囊实性,有包膜(图1B),与周围组织粘连固定,右侧腋动静脉、右锁骨下动静脉受压变形,臂丛神经轻度受压。分离并保护好腋动静脉、右锁骨下动静脉及臂丛神经,沿肿块边缘将肿块完整切除,冲洗术区,彻底止血,右腋顶放置引流管接高负压引流瓶,关闭切口。
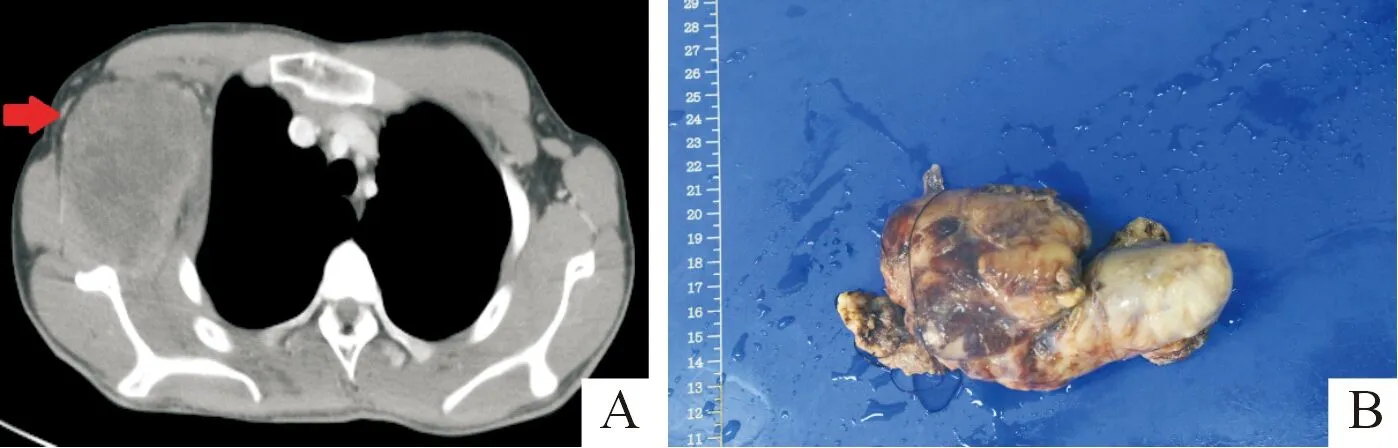
A:患者胸部增强CT,箭头所示右腋窝团块状软组织影,内密度欠均匀,可见不均匀轻度强化;B:大体标本,肿块大小约13 cm×6 cm×5 cm,呈囊实性,有包膜
1.3组织病理学和免疫组化结果见图2。术后病理证实为MTT。镜下见瘤组织主要由致密性梭形细胞组成,伴有大小不等的囊腔,未见明确内衬上皮,核仁明显,可见病理性核分裂,见横纹肌分化。S-100蛋白、结蛋白Desmin和肌原蛋白Myogenin免疫组化染色阳性(图2B~D)。
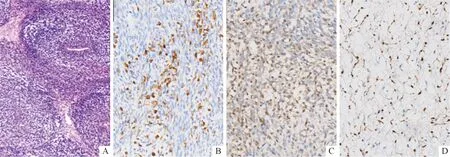
A:肿瘤组织主要由致密性梭形细胞组成,伴有大小不等之囊腔,未见明确内衬上皮,核仁明显,可见病理性核分裂,见横纹肌分化(HE);B:Desmin(+);C:Myogenin(+);D:S-100(+)
2 讨论
MTT是一种罕见的侵袭性肿瘤,在世界卫生组织软组织肿瘤分类(1992年)中,将MTT列为MPNST的一个亚型[14],其起源于神经鞘,伴横纹肌肉瘤样分化,预后不良[15]。免疫组化染色有助于确定肿瘤细胞的来源。S-100蛋白阳性是神经鞘分化的标志,而结蛋白Desmin、肌动蛋白Actin和肌原蛋白Myogenin阳性是横纹肌细胞分化的标志,以上是诊断的关键[16]。本病例S-100蛋白、结蛋白Desmin、肌原蛋白Myogenin均呈阳性,表明肿瘤中含有神经鞘和横纹肌母细胞成分。Daimaru等[17]将MTT的定义扩大到包括散发病例。从流行病学角度来看,70%的MTT病例与Ⅰ型神经纤维瘤病(NF-1)相关[18],其余为偶发性或放疗后出现的病例[19-21]。Brooks等[18]的报告显示,该疾病性别分布均等;Aldalyami等[22]和Rekhi等[23]则描述了该病更多见于男性。大多数病例都是在中年时期(30~50岁)被诊断[15]。至于位置,MTT通常出现在头、颈、躯干和四肢,很少出现在中枢神经系统、脊柱和心脏内[15]。
目前MTT的病因和发病机制还不明确。一种假说认为,恶性施万细胞可分化为横纹肌母细胞[24];而另一种假说认为,这两种细胞均起源于分化程度较低的具有外胚层和中胚层潜能的神经嵴细胞[25]。Nikitin等[26]在大鼠模型中发现,神经鞘瘤细胞能够直接转化为横纹肌母细胞。最近的细胞遗传学研究发现了染色体异常:例如11p15异常可能是横纹肌细胞分化的原因[3];p53去调控也可能在MTT的发生中起作用[27];一些病例可以观测到e-myc致癌基因的扩增[28],这可能是该肿瘤侵袭性行为的原因;Velagaleti等[29]认为定位于9p染色体上的基因改变可能是MTT发生、发展中的早期事件;McComb等[30]在MTT细胞遗传学分析研究中发现有22号染色体的丢失。文献[31]报道,有约8%的MTT患者曾遭受过辐射,这是公认的危险因素。此外,即使是治疗性照射也可能导致MTT:有文献[19]报道了放射治疗后良性神经鞘瘤向MTT的恶性进展。
MTT的主要临床表现为进行性增大的肿块及其引起的神经症状。CT或MRI仅能对肿块定位而无法定性,故难以早期诊断。MTT和MPNST的影像表现缺乏特征性,二者均表现为巨大的软组织肿块,但MTT伴有异源成分更多,恶性程度更高,肿块内囊变、坏死及出血的影像表现更为常见。且MTT通常大于MPNST,其最大截面平均为9.0 cm, MPNST为6.0 cm[31-32]。
目前还没有治疗MTT的指南,公认的最佳治疗方案是根治性切除[5],手术切除后的高剂量放射治疗也被广泛接受。McConnell等[31]报道,手术完全切除后的放疗可提高患者的生存率,但与减少复发或进展无关。截至目前,没有证据表明化疗有效;然而,化疗方案,如PEI(顺铂、依托泊苷、异环磷酰胺)作为一线化疗方案和IA(异环磷酰胺、阿霉素)或MAID(美西娜、阿霉素、异环磷酰胺和达卡巴嗪)已被提议作为二线治疗[33]。在肿瘤标本中存在视黄酸受体的情况下,也报道了对异维甲酸和干扰素α治疗的有利反应[34]。
MTT侵袭性强,预后极差,其5 a生存率只有14%,中位死亡时间为13个月,约1/3的患者出现远处转移,50%的患者出现局部进展或复发,复发/进展的中位时间为6个月[31]。其预后与NF-1、年龄、大小、手术是否完全切除、组织学检查是否存在切缘受侵以及肿瘤在主干中的位置有关[15, 31]。
