天冬酰胺合成酶缺乏症1 例报告并文献复习
2021-02-26索桂海汤继宏张兵兵王曼丽
索桂海 汤继宏 冯 隽 张兵兵 王曼丽
1.苏州大学附属儿童医院(江苏苏州 215003);2.南通大学附属医院儿科(江苏南通 226001)
天冬酰胺合成酶缺乏症(asparagine synthetase deficiency,ASNSD)是一种罕见的常染色体隐性遗传的先天性代谢缺陷病,为染色体7q21上的ASNS基因变异所致,可导致严重的神经系统疾病,其临床特征为小头畸形、严重的精神运动发育迟缓、渐进性脑病、皮质萎缩、难治性癫痫发作,部分患儿可出现喂养困难、呼吸功能不全等。头颅磁共振(MRⅠ)显示小头畸形,脑萎缩,髓鞘形化延迟,脑回形态简化。ASNSD十分罕见,发病机制尚不清楚。由于无特异性的生化指标及临床表现,目前报道的所有病例均是通过全外显子测序确诊。本文回顾分析1 例ASNSD 患儿的临床资料及全外显子组测序结果。
1 临床资料
患儿,女性,首次来苏州大学附属儿童医院就诊年龄为5岁3月龄。患儿8月龄出现惊癫痫发作,为全面性强直、阵挛发作,约半个月发作1次。头颅磁共振(MRⅠ)检查未见异常。脑电图可见棘波、棘慢波发放。外院诊断为癫痫,先后予丙戊酸钠、托吡酯、左乙拉西坦联合治疗,但仍有发作。患儿系G1P1,胎龄39+5周,自然产道分娩,出生体质量3 100 g,头围、身长不详,无窒息抢救病史,母孕期未见异常。患儿出生后7 月龄会抬头,12 月龄独坐,18 月龄扶站,18 月龄能无意识发“爸爸”、“妈妈”等复音,24月龄后渐出现生长发育倒退。就诊时5岁3月龄,不能抬头、不能坐、不会爬、不能站、不能走,不会抓握,不能发音,与人无眼神交流。患儿父母无异常表现。此后患儿母亲怀孕2 次均流产,原因不详。家族中无类似疾病史。体格检查:体质量16 kg(<-1 SD),身高108 cm(<-1 SD),头围44 cm(<-3SD);神志清,呼吸平稳,巨耳,双眼睑无下垂,瞳孔对光反射灵敏,眼球活动正常,双侧鼻唇沟对称,颈软,四肢肌力Ⅱ级,肌张力增高,腱反射亢进。实验室检查:血常规、肝肾功能、电解质、心肌酶谱、血氨、血乳酸等均未见异常;血和尿遗传代谢病筛查未见异常。头颅MRⅠ示幕上脑室扩张,脑沟及脑裂广泛增宽加深,脑白质偏少,胼胝体菲薄(图1)。
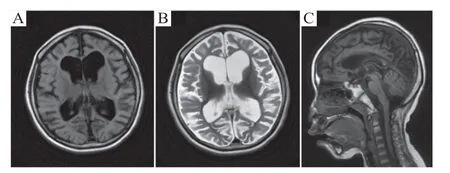
图1 患儿头颅MRI 表现
为进一步明确病因,经患儿家属知情同意及苏州大学附属儿童医院医学伦理委员会审核批准后,委托北京迈基诺基因科技有限责任公司行外周血基因检测。全外显子测序发现,患儿ASNS基因存在复合杂合变异,1 个为整码变异c.1503_1505 delAGC,导致氨基酸改变p.501_502 delAAinsA,源自父亲;另1 个为错义变异c.776 G>C,导致氨基酸改变p.G259A,源自母亲(图2)。线粒体环基因panel未见异常;全基因组拷贝数变异(copy number variations,CNV)分析未见异常。利用SWⅠSSMODEL(https://swissmodel.expasy.org/),以6 gq 3.1 A 为模版进行同源预测建模(QMQE为0.96,QMWAN为-0.34,seq identity 为100%)。黄色球状位置为501-502 AA,红色球状位置为259 G。与野生型蛋白相比,变异基因改变了蛋白结构(图3)。生物信息学软件(Provean、mutationtaster)均预测其致病。基因学检测结合临床表现患儿确诊为ASNSD。本例患儿的整码变异及错义变异为新发变异,在HGMD 数据库未有该位点的相关报道,查阅既往文献及数据库未见该位点的相关报道。
患儿首次就诊后,予调整抗癫痫药物剂量(丙戊酸钠33 mg/kg,托吡酯4 mg/kg,左乙拉西坦40 mg/kg),同时予康复治疗。半年后门诊随访,癫痫发作仍未控制,精神运动发育迟缓无改善。
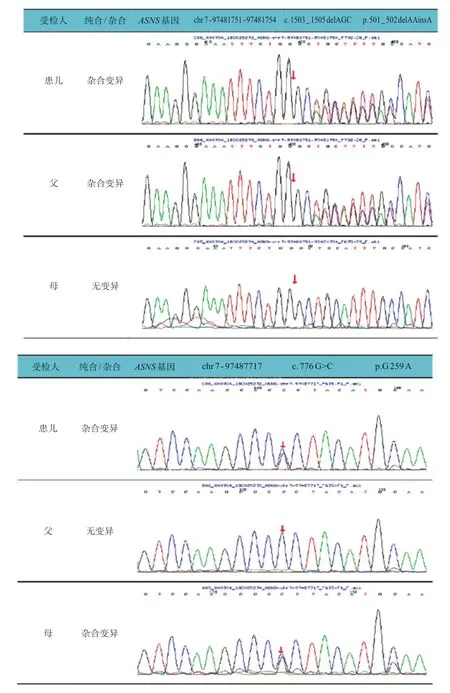
图2 患儿及其父母ASNS 基因测序图
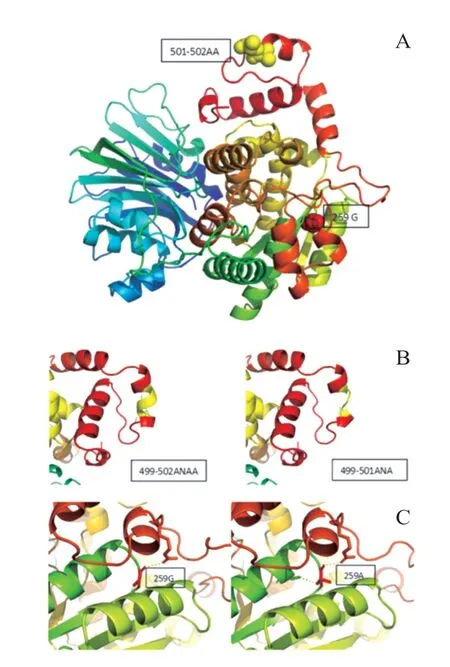
图3 蛋白三级结构图
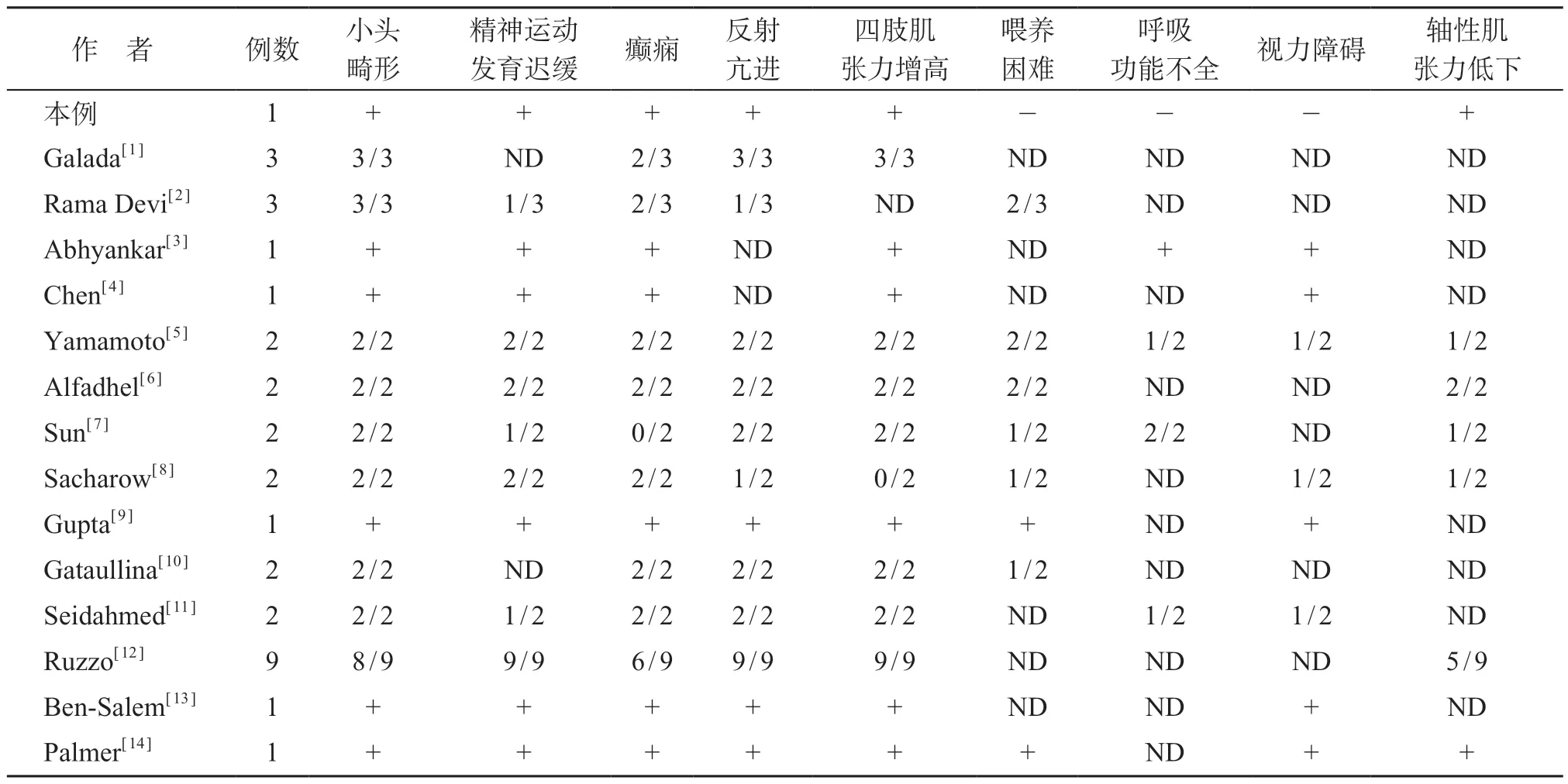
表1 33例天冬酰胺合成酶缺乏症患儿临床特征总结
2 讨论
通过对关键词“ASNSD、ASNS”的文献搜索,发现包括本例患儿,迄今共报告33例ASNSD,其临床特征见表1。33例患儿中,32例存在小头畸形,24例精神运动发育迟缓,26 例癫痫,27 例四肢肌张力增高,27例反射亢进。小头畸形、精神运动发育迟缓、癫痫及四肢肌张力增高、反射亢进是该病的重要特征,部分患儿可出现喂养困难、呼吸功能不全、轴性肌张力低下、视力障碍、额头倾斜、惊跳症等,少数患儿可合并膈膨出、小下颌畸形、巨耳畸形等。ASNSD 患儿癫痫发作可呈现多种形式,包括肌阵挛、强直、痉挛、全身性强直-阵挛发作,且多为难治性癫痫。有报道大剂量丙戊酸钠 [80 mg/(kg·d)]有助于控制全面性强直阵挛发作和肌阵挛发作,但对局灶性意识障碍发作无效[15]。由于存在喂养困难和呼吸功能不全,多数患儿在婴儿早期死亡[12]。ASNSD患儿头颅MRⅠ可出现脑容量减少,脑回简单化,巨脑室,髓鞘化延迟及脑桥、胼胝体及皮质发育不良等。本例患儿8月龄出现全身性强直发作、阵挛发作,予多种抗癫痫药物联合治疗近5 年仍未控制,现5岁3月余,头围44 cm,生长发育倒退,不能坐、不能站、不会爬、不能走,不会抓握,不能发音,与人无眼神交流,头颅MRⅠ提示幕上脑室扩张、脑萎缩、胼胝体菲薄,均符合ASNSD综合征临床特征。
ASNSD 是一种罕见的常染色体隐性遗传病,患病率不足1/ 100万[11],为染色体7q21上的ASNS基因变异所致。ASNS基因全长35 kb,包含l3个外显子,有3种转录剪切方式,至少产生3 种转录本,但只有1 种可读框,最终编码产生1 种酶蛋白,即天冬酰胺合成酶。ASNS 属于氨基酸转移酶家族[16],相对分子质量约为65 000,包含561个氨基酸,在人体器官中有不同程度的表达,脑内ASNS 表达水平较高,对脑的形成至关重要[12],但脑脊液中的ASNS水平却显著降低[17]。该酶催化合成天冬酰胺和谷氨酸。天冬酰胺是非必须氨基酸,细胞既能从外界摄取又能内源性合成,因此ASNS缺乏或功能障碍导致的严重组织特异性表型的确切机制尚不清楚[9,12,14]。ASNS缺陷引起神经功能障碍的机制可能为ASNS缺乏导致天冬酰胺消耗引起神经元凋亡增加,导致脑萎缩、神经元兴奋性增高,进而导致天冬酰胺和谷氨酸在脑中积累引起癫痫发作和惊跳症[9]。
ASNSD 无特异性生化指标,虽然有研究发现部分患者血清及脑脊液中的天冬酰胺、谷氨酰胺和天冬氨酸水平出现异常,但是上述指标正常不能排除该病,相对于血清而言,脑脊液中天冬酰胺水平降低可能更有助于疾病的诊断[7]。目前ASNSD 的确诊仍依赖于全外显子测序。本例患儿ASNS基因存在复合杂合变异,一个是整码变异c.1503_1505delAGC,导致氨基酸改变p.501_502delAAinsA,源自父亲,生物信息学蛋白功能预测软件SⅠFT、PolyPhen_2、REVEL预测均为未知;一个是错义变异c.776 G>C,导致氨基酸改变p.G259A,源自母亲。结合临床表现患儿诊断为ASNSD。到目前为止,已经报道ASNS基因26种不同变异,包括23 种错义变异,3种无义变异(3种移码变异和1种点突变)。
ASNSD预后很差,除支持性治疗,尚无有效治疗方法。曾有报道尝试补充天冬酰胺治疗,且补充天冬酰胺后患儿精神状态较前改善,从植物人状态略微改善到最低意识状态,但随着治疗继续患儿易怒、睡眠障碍,并且癫痫发作恶化,导致治疗中断[15]。因此这种干预措施是否有效尚需要更多的研究来确定。
综上所述,ASNSD是ASNS基因突变导致的常染色体隐性遗传病,任何出现小头畸形、严重的精神运动发育迟缓、难治性癫痫发作等表现的患儿都应该考虑ASNSD可能。到目前为止,所有的ASNSD病例都是通过外显子组测序而确定,因此疑似ASNSD 的患儿应尽早行外显子组测序,以明确诊断。
