加替沙星对映异构体拆分研究*
2021-02-25鄢雷娜刘绪平段和祥陈伟康
陈 希,鄢雷娜,刘绪平,段和祥,陈伟康
(江西省药品检验检测研究院,国家药品监督管理局中成药质量评价重点实验室,江西省药品与医疗器械质量工程技术研究中心,江西 南昌330029)
当前对映异构体(对映体)的拆分方法主要包括高效液相色谱法(HPLC)、毛细管电泳法(CE),其中HPLC 法又分为柱前衍生法、手性流动相添加法和手性固定相法[1-6]。柱前衍生法前处理复杂且时间长;以CuSO4和手性氨基酸为手性流动相的方法已普遍用于喹诺酮类药物对映体的拆分,原理为待测物可与Cu2+及手性氨基酸形成三元非对映体配位化合物而分离,该法适用于手性碳原子与手性配合部位距离较近的化合物分离,如氧氟沙星、帕珠沙星等,但对于手性碳与手性配合部位距离较远的药物却无法拆分,如手性碳在哌嗪侧链上的洛美沙星和加替沙星,熊正平等尝试采用过L-脯氨酸与CuSO4的手性流动相添加剂法来拆分洛美沙星对映体未能成功[7-9]。
手性固定相法利用药物对映体和固定相之间所形成的非对映体复合物稳定性差异而实现对映体的拆分,虽然手性色谱柱较贵,但优点是适用于不含活泼反应基团的化合物,且样品处理步骤简单,现已广泛应用在药物对映体拆分上[10,11]。本文利用以键合了苯基氨基甲酸酯化β-环糊精的手性色谱柱(Chiral CD-Ph)对加替沙星对映体进行了拆分研究。

图1 加替沙星的化学结构式Fig.1 Chemical structure of gatifloxacin
1 材料
1.1 仪器
岛津LC-AD 高效液相色谱仪,METTLER MS205DU 电子天平。
1.2 药品与试剂
加替沙星对照品(来源:中国食品药品检定研究院,批号:130518-200402,含量:97.2%;旋光度测定为0°,认为左右旋加替沙星比例为1∶1);加替沙星片(湖北潜江制药股份有限公司,批号180801);水为纯化水,HClO4和NaClO4为分析纯,甲醇、乙腈为色谱纯。
2 方法与结果
2.1 色谱条件
流动相:0.5mol·L-1NaClO4溶液(用HClO4调节pH 值至4.0)-甲醇(53∶47);流速:1.0mL·min-1;色谱柱为Chiral CD-Ph(4.6mm×250mm,5μm);柱温40℃;进样量10μL;检测波长:292nm。
2.2 溶液的制备
精密称取加替沙星对照品适量,加50%的甲醇溶解并稀释成含加替沙星约为2mg·mL-1的对照品溶液。取加替沙星片研细,精密称取适量,用50%的甲醇溶解并稀释成含加替沙星约为1mg·mL-1的供试品溶液。
2.3 盐和有机相的选择
分别以甲酸铵、KH2PO4和NaClO4作为流动相对加替沙星进行拆分,结果显示甲酸铵和KH2PO4系统均无法拆分加替沙星,而NaClO4系统有拆分加替沙星的可能。以NaClO4为水相,分别以甲醇和乙腈为有机相对加替沙星进行拆分,结果显示乙腈拆分效果很差,几乎无法拆分加替沙星,而甲醇拆分效果较好,最终选择甲醇进行研究。
2.4 NaClO4 浓度的选择
以NaClO4-甲醇为流动相,尝试NaClO4浓度为0.1、0.3 和0.5mol·L-1时对拆分结果的影响,结果两对映体间分离度分别为1.092、1.483 和1.602,最终选择浓度为0.5mol·L-1的HClO4进行研究。
2.5 甲醇比例的选择
甲醇50%时对映体保留时间约为34min,对映体间分离度1.498;加大甲醇比例两对映体间分离度更小,减少比例分离度有加大趋势,但保留时间太长,综合考虑选择甲醇比例为47%进行研究。
2.6 系统适应性实验
对照品溶液,按“2.1”中色谱条件进行测定,色谱图详见图2。图谱显示加替沙星两个对映体间保留时间分别约为34 和39min,两对映体峰间的分离度为1.6,符合《中国药典》2015 年中待测物质色谱峰间的分离度应大于1.5 的要求[12]。

图2 加替沙星对照品色谱图Fig.2 Gatifloxacin reference chromatogram
2.7 线性关系考察
取对照品溶液适量,分别加50%甲醇分别稀释成含加替沙星100、250、500、750、1000 和1500μg·mL-1的溶液(单个对映体浓度为加替沙星浓度的50%),按“2.1”中色谱条件进行测定,记录色谱图。以加替沙星单个对映体的峰面积为纵坐标(y),加替沙星单个对映体的浓度为横坐标(x)绘制标准曲线。结果表明,加替沙星单个对映体浓度在50~750μg·mL-1之间与相应峰面积呈良好线性关系。对映体1 回归方程为:y=52819x+44509,r2=0.9999;对映体2 回归方程为:y=56850x+404273,r2=0.9999。
2.8 精密度和重复性
取加替沙星浓度约为1mg·mL-1的对照品溶液,按“2.1”中色谱条件连续进样6 针,记录色谱图。按“2.2”中方法分别制备6 份供试品溶液,按“2.1”中色谱条件分别进样,记录色谱图。分别计算加替沙星每个对映体峰面积的RSD,结果均小于1%,说明本方法精密度和重复性良好。
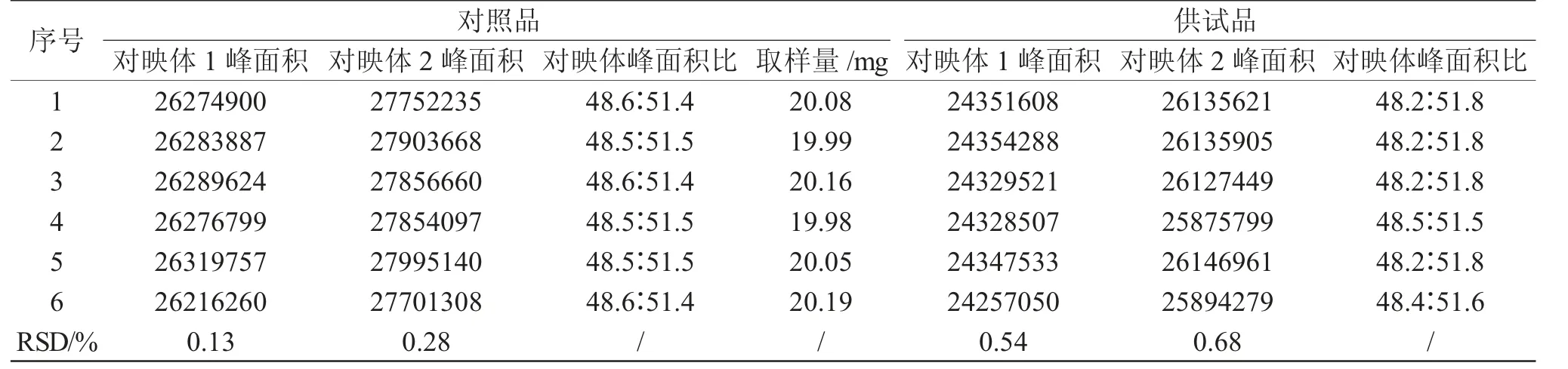
表2 精密度和重复性测定结果Tab.2 Results of precision and repeatability
2.9 定量限与检出限考察
取对照品溶液倍比稀释,按“2.1”中色谱条件测定,以信噪比满足3∶1 和10∶1 计算检出限和定量限。结果本方法加替沙星单个对映体的检出限和定量限分别为1.5 和5μg·mL-1。
2.10 样品测定
取供试品溶液按“2.1”中色谱条件测定,记录色谱图。结果供试品中对映体1 和对映体2 峰面积比值平均为48.3∶51.7,与对照品溶液对映体峰面积比接近,表明供试品溶液中两对映体含量近似一致。
3 结论
本研究使用手性色谱柱Chiral CD-Ph 成功拆分了加替沙星对映体,以0.5mol·L-1NaClO4溶液(pH 值4.0)-甲醇(53∶47)为流动相时,加替沙星两个对映异构体分离度最佳,解决了手性流动相方法无法拆分加替沙星的难题。该方法样品前处理简单,精密度和重复性良好、灵敏度高,可用于加替沙星光学纯度的测定和质量控制。
