Mn掺杂LiNbO3结构ZnTiO3的磁性和光电性质的第一性原理研究
2021-02-23苏锟仁梁一机林尔庆徐祥福陈星源赖国霞
苏锟仁,梁一机,林尔庆,王 国,徐祥福,陈星源,赖国霞
(广东石油化工学院理学院,物理实验教学中心,茂名 525000)
0 引 言
极性化合物具有铁电性、压电性或二阶非线性光学等特性,在物理学和材料科学领域引起了广泛的关注[1-2]。LiNbO3型结构钙钛矿化合物是一种典型的极性化合物[3]。通常大多数的LiNbO3型化合物不能在一般条件下合成,需要高温和高压的条件辅助[4]。Inaguma等[5]在2014年通过高温高压的方法合成了一种新的极性化合物LiNbO3结构的ZnTiO3(LN-ZnTiO3)。Ruiz-Fuertes等[6]通过拉曼光谱和二次谐波等测试,发现LN-ZnTiO3具有铁电性,但是随着压强增大到20 GPa会发生顺电相的相变。Zhang等[7]通过密度泛函理论计算了LN-ZnTiO3的电子结构和光学性质,发现LN-ZnTiO3是一种宽带隙半导体,同时具有较高的压电系数和非线性光学系数。事实上目前应用比较广泛的是钛铁矿(IL)型结构的ZnTiO3材料,其在微波电介质、气体传感器和太阳能电池中都有实际应用[8-10]。关于ZnTiO3材料在磁学的应用方面的报道还比较少。最近Yan等在硅晶片衬底上制备了Co掺杂IL-ZnTiO3纳米晶体膜,发现Co掺杂不仅可以在IL-ZnTiO3中提供磁性,还可以降低材料的带隙,拓展了材料在磁光器件方面的应用[11]。Wattanawikkam等[12]通过超声化学法制备了Mn掺杂IL-ZnTiO3的样品,有助于增强光催化性能。在本文中,计算了Mn离子掺杂LN-ZnTiO3的磁性和光吸收性质,发现Mn离子掺杂可以在LN-ZnTiO3中产生较大的局域磁矩,降低带隙,促进可见光的吸收。
1 计算方法

图1 LN-ZnTiO3的结构图
本文的计算工作主要采用基于密度泛函理论赝势平面波VASP软件包完成[13]。计算的平面波截断能取为500 eV,交换关联势选取PBE方法[14],赝势选取的是投影平面波(PAW)方法。布里渊区内的K点取样为0.3 nm-1。对于3 d过渡金属Mn元素采取了Hubbard U的修正方法[15],其中U值参考铁电材料中的典型数值取为4 eV[16-17]。一般PBE的方法会低估材料的带隙,本文也采取了Modified Becke-Johnson (MBJ)势方法[18-19]计算电子结构和带隙。计算的精度设置为每个电子步的能量小于10-5eV,力的收敛判据为小于0.2 eV/nm。如图1所示,计算的LN-ZnTiO3结构共30个原子。
根据文献[12]报道,Mn掺杂3%、5%、7%(摩尔分数)时性质变化的趋势是相同的,为与实验相符,同时为了计算方便,本文中计算模型所采用的掺杂浓度设为3.33%(摩尔分数)。为了通过理论计算确定掺杂的替代位置,同时计算了Mn原子替代掺杂LN-ZnTiO3材料中Zn离子或Ti离子的结构。计算的晶格参数如表1所示,其中计算的本征LN-ZnTiO3的晶格参数和实验值[5]比较接近。

表1 LN-ZnTiO3晶格参数
2 结果与讨论
2.1 掺杂稳定性的分析
Mn替代掺杂LN-ZnTiO3材料中的Zn离子或Ti离子取决于材料的掺杂形成能。采取如下的形成能公式(1):
E1=E(Zn5MnTi6O18)+E(Zn)-E(Zn6Ti6O18)-E(Mn)E2=E(Zn6Ti5MnO18)+E(Ti)-E(Zn6Ti6O18)-E(Mn)
(1)
其中E1和E2分别为Mn替代掺杂Zn以及Mn替代掺杂Ti的形成能。E(Zn5MnTi6O18)和E(Zn6Ti5MnO18)为掺杂体系的总能。E(Zn6Ti6O18)、E(Zn)、E(Mn)和E(Ti)分别是各自对应的本征体相结构的能量。E1、E2的计算结果分别为-1.5 eV、4.4 eV,这说明在Mn掺杂LN-ZnTiO3中,Mn倾向掺杂Zn位,形成能比较低。这与Mn掺杂IL-ZnTiO3的结果类似,在实验中发现了Mn掺杂IL-ZnTiO3主要是替代Zn位[12]。在LN-ZnTiO3中,Zn离子为+2价3d10的电子构型,而Ti离子为+4价3d0的电子构型。Mn离子替代Zn离子容易形成+2价3d5的电子构型,而掺杂Ti离子则容易形成+4价,电子构型为3d3。根据洪特定则和晶体场理论,Mn的3d5构型容易形成稳定的构型。在LN-ZnTiO3中,Mn掺杂Zn位比掺杂Ti位相对稳定。因此主要分析Zn5MnTi6O18的磁性和光电性质。如表1所示,掺杂后的晶格参数增大,原因是Mn2+离子半径比Zn2+离子半径大。
2.2 磁性分析
如图2(a)(b)所示,本征LN-ZnTiO3和Zn5MnTi6O18的自旋电荷密度图表明在Zn5MnTi6O18中自旋向上和自旋向下的电荷密度并没有相互抵消,自旋电荷主要集中在Mn离子附近。过渡金属Mn离子的掺杂在LN-ZnTiO3中会引起局域磁性的变化。

图2 (a)LN-ZnTiO3和(b)Zn5MnTi6O18的自旋电荷密度图,电荷密度的单位为0.05 e/bohr3
这是因为在LN-ZnTiO3中Zn2+的3d10电子构型和Ti4+的3d0电子构型并不会引起磁性的变化,而Mn2+的3d5电子构型则会引起磁性的变化。在VASP软件包中读取的Mn离子磁矩为5 μB。如图3的分波态密度所示,Mn离子5个d轨道均有自旋向上的电子占据,每个电子分别贡献1 μB磁矩,掺杂体系一共呈现5 μB总磁矩。

图3 Mn原子的分波态密度图
Zn5MnTi6O18的磁矩主要来源于Mn离子的3d轨道。Mn离子3d轨道的态密度峰值主要是在费米能级附近,容易与O离子的p轨道形成明显的杂化交叠,这对Zn5MnTi6O18的物理性质将会产生重要的影响。
2.3 光电性质的分析
能带结构对材料的光电性质起着重要的作用。到目前为止,还没有关于LN-ZnTiO3带隙的实验报道。如图4(a)所示,本文采取了MBJ的方法计算了本征LN-ZnTiO3的能带结构为间接带隙结构,得到带隙的数值为3.1 eV。计算得到LN-ZnTiO3带隙为3.1 eV,表明材料很少吸收可见光。Zhang等[7]采取模守恒赝势方法计算得到的LN-ZnTiO3带隙为3.2 eV。本文的计算结果与Zhang等的计算结果很接近。由图4(b)可知本征LN-ZnTiO3的价带顶主要由O-2p轨道组成,而导带底由Ti-3d轨道组成。

图4 LN-ZnTiO3 能带图(a)和态密度图(b);Zn5MnTi6O18的能带图(c)和态密度图(d)
计算的Zn5MnTi6O18的能带结构如图4(c)所示。从图中可以看出,掺杂以后,在费米能级以下,禁带中增多了2个电子态,位置靠近价带。由图4(d)可以看出,导带底主要由Ti-3d电子贡献,价带顶是由O-2p轨道贡献,这符合ABO3材料的一般特性[20]。根据图4(d)DOS态密度图,此电子态是由O-2p轨道与M-3d轨道杂化形成的,结合前述图2,Mn掺杂LN-ZnTiO3产生磁性原因是O-2p轨道与Mn-3d的共价杂化,这也是ABO3材料铁电性质一般来源[20-21],验证了本计算符合此类材料的一般规律。由图4(c)亦可以看出,掺杂Mn后,形成的缺陷能级位于价带上方,虽然是同价掺杂,但由于Mn与Zn原子半径不同,造成了晶格畸变,形成结构缺陷,在禁带中引入缺陷能级,形成P型掺杂。缺陷能级的引入减小了可见光范围的光吸收阈值,光吸收阈值数值变为2.4 eV。
通过能带图(见图4(c))和 态密度(见图4(d))可知由于在掺杂位置缺陷局域态的存在,光吸收时,由缺陷能级跃迁至导带,跃迁能量变低,另外由于此缺陷能级为浅能级,增加了非平衡载流子的产生概率,基于这两个原因,容易发生在可见光范围的Mn-3d和O-2p向Ti-3d轨道的跃迁,促进了可见光的吸收。为了进一步比较对可见光的吸收情况,计算了LN-ZnTiO3和Zn5MnTi6O18的吸收光谱,计算公式(2)如下所示:
(2)
其中:I为光学吸收系数;ε1为介电函数实部;ε2为介电函数虚部;ω为入射光的频率。
图5为LN-ZnTiO3和Zn5MnTi6O18的光吸收谱。从图中可以看出,Zn5MnTi6O18在可见光范围内比LN-ZnTiO3更容易形成明显的小峰,吸收明显增强,与前文分析结果相符。
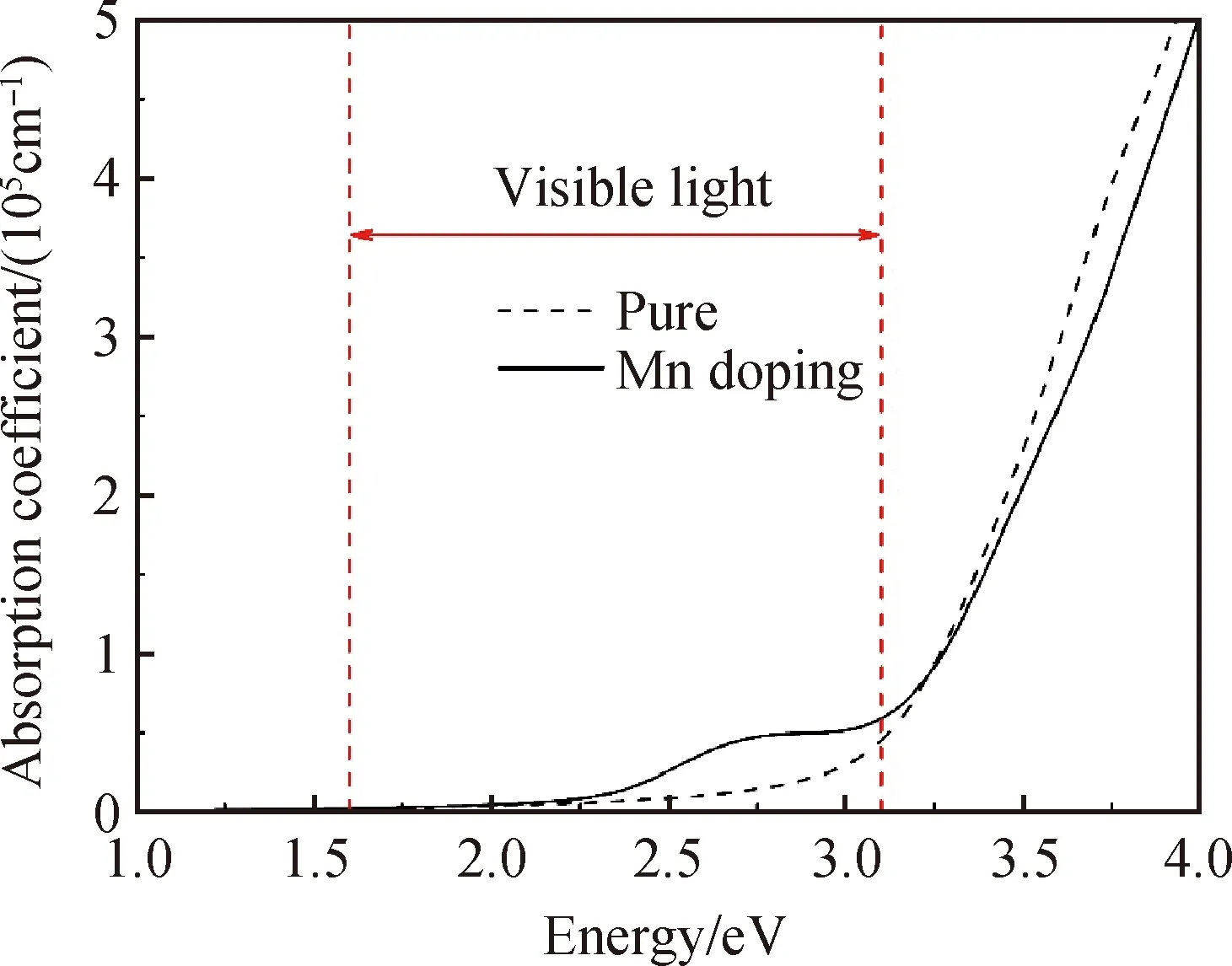
图5 本征和Mn掺杂LN-ZnTiO3的光吸收谱
3 结 论
基于密度泛函理论,利用VASP软件包计算了Mn掺杂LN-ZnTiO3的结构、磁性和光电性质。计算结果表明Mn掺杂LN-ZnTiO3倾向占据Zn位,晶格参数有所增大。态密度分析表明Mn替代Zn位掺杂形成了稳定的3d5电子构型,可以为材料提供较大的局域磁矩,约为5 μB。Mn掺杂LN-ZnTiO3会在价带顶附近形成明显的Mn-3d和O-2p轨道组成的受主能级,降低材料的光吸收阈值,促进可见光的吸收。在LN-ZnTiO3中掺杂Mn可以同时实现较大的局域磁矩和p型半导体的特性,可以促进材料在磁学和可见光吸收领域的应用。
