湿法糖基化处理大豆11S蛋白后的表面活性变化
2021-02-06杜沁岭周思懿吴岱泽张效铭杨文钰
杜沁岭 周思懿 吴岱泽 张效铭 王 珮 杨文钰 张 清
(四川农业大学食品学院1,雅安 625014) (四川省作物带状复合种植工程技术研究中心2,成都 611130)
大豆11S蛋白是大豆蛋白的主要储藏蛋白之一,约占大豆蛋白总量的31%[1],与大豆蛋白的功能性质密切相关[2]。大豆11S蛋白具有水合性质、乳化性、持水持油性等表面活性,同时还具有可降解性,是一种环境友好的天然可再生资源。然而,由于提取的大豆 11S 蛋白具有紧密的分子结构,多数基团被包裹在分子内部,导致其本身表面活性较差,在食品中的应用受到限制[3]。所以,为了改良大豆11S蛋白的表面活性,需要对其进行改性处理。
糖基化改性具有反应过程温和、改性蛋白性质稳定和无有毒有害试剂添加等优点,常被选为大豆蛋白的理想改性方法[4]。大豆蛋白的糖基化改性已有广泛研究,潘男等[5]、Lu等[6]和Seo等[7]分别研究了大豆蛋白与不同种类多糖发生糖基化反应后凝胶性、溶解性、乳化性等功能性质的改善情况。另外,前人也对大豆11S蛋白的二级结构、表面疏水性[8]、热处理凝胶性[9]进行了研究。而对于大豆11S蛋白与多糖发生糖基化反应改性后表面活性变化的研究却鲜有报道。
本研究以3种不同分子质量大小(4、10、70 ku)的葡聚糖对大豆11S蛋白进行湿法糖基化反应,通过十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)和荧光光谱测定其反应发生程度,对大豆11S蛋白-葡聚糖轭合物的表面疏水性、Zeta电位、溶解性(不同pH条件下)、乳化性、持水持油性进行测定,从结构特征和功能性质两方面分析糖基化反应后表面活性变化。
1 材料与方法
1.1 材料与试剂
大豆(合丰57),葡聚糖(4、10、70 ku);Lowry法蛋白质含量测定试剂盒,8-苯胺萘磺-1-酸盐(ANS),大豆油,其他化学试剂均为分析纯级。
1.2 主要仪器与设备
高速粉碎机(CS-700),台式恒温振荡器(150638),高速剪切均质机(AD500S-H),冷冻干燥机(FD-1A-50),冷冻离心机(Micro17),紫外-可见分光光度计(UV-6),荧光分光光度计(Thermo scientific R),Zeta电位仪(Nano ZS)。
1.3 试验方法
1.3.1 大豆11S蛋白的提取
大豆经粉碎机粉碎1 min过80目筛后置于锥形瓶中,与乙醚(1∶8)混匀后振荡4 h(25 ℃,110 r/min);静置15 min,待料液分层后弃去上层溶剂,将下层脱脂大豆粉置于通风橱中风干[9]后分散在0.02 mol/L的磷酸盐缓冲液中(1∶10),在45 ℃、pH 8.5的条件下磁力搅拌1 h后离心(4 ℃,8 500 r/min,20 min)。向上清液中加入NaHSO3使其浓度为0.01 mol/L,调节pH至6.4;于4 ℃冰箱过夜后再离心(4 ℃,6 500 r/min,20 min),沉淀部分即为11S蛋白[10]。通过SDS-PAGE对提取样品进行鉴定。
1.3.2 大豆11S蛋白湿法糖基化处理
将大豆11S蛋白配成5 mg/mL的蛋白溶液,再加入等质量的葡聚糖,混匀后密封放置在80℃的恒温水浴锅中进行糖基化反应,分别反应1、2、3、4、5 h后取出样品,迅速放入冰水浴中终止反应,得到大豆11S蛋白-葡聚糖轭合物。将样品冷冻干燥后置于4 ℃条件下保存备用。
1.3.3 SDS-PAGE测定
分离胶、浓缩胶浓度分别为13%、5%,样品质量浓度为2 mg/mL,上样量为10 μL,凝胶电泳于恒定电压下进行(120 V)。电泳后用考马斯亮蓝R-250染色,脱色完成后,对凝胶拍照并进行分析[11]。
1.3.4 内源荧光光谱测定
样品分散在10 mmol/L磷酸盐缓冲液(pH=7.0)中,浓度为0.15 mg/mL,设置激发波长为290 nm(狭缝宽度5 nm),以20 nm/s的速率在300~510 nm内扫描,测定荧光强度[12]。
1.3.5 表面疏水性测定
将糖基化蛋白样品分散于蒸馏水中制成蛋白浓度1 mg/mL的溶液,充分搅拌后离心(4 ℃,10 220r/min,20 min),取上清液4 mL,加入8 mmol/L的ANS溶液40 μL,充分振荡混匀后静置3 min,测定样品的荧光强度(FI)。设置激发波长为365 nm(狭缝宽度5 nm),以20 nm/s的速率在400~600 nm范围内扫描,测定荧光强度。以扫描范围内最大荧光强度值作为蛋白质的表面疏水性(H0)[13]。
1.3.6 Zeta电位测定
配制0.02 mol/L Tris-HCl缓冲液(pH 7.0),脱气后,再加入糖基化产物充分溶解至蛋白质量分数为0.2%,测定zeta电位,测定温度为25 ℃,重复测定3次以上,取平均值[14]。
1.3.7 不同pH条件下溶解性测定
将10 mg糖基化蛋白样品分散于10 mL蒸馏水中,混匀后用0.05 mol/L乙酸溶液、0.05 mol/L磷酸盐溶液、0.05 mol/L Tris溶液分别调节溶液pH至pH=3、4和5;pH=6、7和8;pH=9。配置好后离心(4 ℃,10 220 r/min,20 min),采用Lowry法测定上清液中蛋白质含量,以0.5 mg/mL牛血清白蛋白为标准物制作标准曲线(标准曲线方程为:y=47.509x+0.066 1,R2=0.998 2)。蛋白质溶解度表示为上清液中蛋白质量与样品中总蛋白质量的比值[15]。

(1)
式中:m1为上清液中蛋白质量;m0为样品中总蛋白质量。
1.3.8 乳化活性(EA)及乳化稳定性(ES)测定
将糖基化蛋白样品分散于pH=6.5、0.1 mol/L磷酸盐缓冲液中制成蛋白浓度为1 mg/mL溶液,按体积比4∶1加入大豆油,于2 000 r/min下高速匀浆1 min,立即或10 min后从距烧杯底部0.5 cm处取匀浆液100 μL加入到10 mL 0.1%的SDS溶液中,振荡混匀后用紫外分光光度计在500 nm波长处测定吸光度,记作A0或A10。以0.1%的SDS溶液作空白对照。蛋白溶液的EA和ES,分别用式(2)、式(3)计算[16]。

(2)
(3)
式中:c为蛋白质质量浓度/g/mL;φ为油相所占体积分数(φ=0.25);A10为静置10 min时乳状液的吸光值;A0为静置0 min时乳状液的吸光值。
1.3.9 持水性(WRC)及持油性(ORC)测定
将50 mg(m0)糖基化蛋白样品分散于10 mL蒸馏水中,室温下水合18 h后离心(4 ℃,10 000r/min,20 min),弃去上清液,记录沉淀质量m1,再将沉淀在60 ℃下干燥至恒重,记录质量m2。在相同操作步骤下,用大豆油代替蒸馏水即为持油性测定方法。按照式(4)计算 WRC或ORC[17]。
(4)
1.4 统计分析
每个样品重复3次实验;采用差异显著性分析处理数据,并由SPSS V17.0软件完成,P<0.05为显著性差异;采用Origin9.0软件作图。
2 结果与分析
2.1 大豆11S蛋白湿法糖基化反应程度分析
2.1.1 SDS-PAGE电泳图谱分析
如图1所示,与marker相比,泳道1中11S蛋白条带颜色明显,其余条带颜色较浅,有少量杂蛋白残余。目前鲜见研究能提取出纯净的11S蛋白,这说明本实验从大豆蛋白中提取出的11S蛋白可作为样品使用。对比泳道1~6,大豆11S蛋白与不同分子质量葡聚糖在发生糖基化反应后,酸性和碱性亚基条带颜色随反应时间延长而逐渐变浅,4 h最浅。考马斯亮兰作为染料常与蛋白质中的自由氨基结合而呈现颜色,因此条带颜色变浅可以间接证明自由氨基减少[18],这表明大豆11S蛋白的氨基与葡聚糖的羰基发生共价结合即发生了糖基化反应且在4 h时反应程度最大。泳道2~6在分离胶顶部出现新的化合物条带,这是因为糖基化反应生成了大分子聚合物,即为蛋白-多糖轭合物,由于其分子质量较大,不能穿过凝胶网络而堆积在凝胶顶部。因此本实验中大豆11S蛋白发生了糖基化反应并产生了大分子物质。该结果与章鼎敏等[19]的分析相似。

注:M为标准分子质量蛋白;1为大豆11S蛋白;2~6分别代表反应1、2、3、4、5 h大豆11S蛋白-葡聚糖轭合物。图1 大豆11S蛋白分别与4、10、70 ku 葡聚糖发生糖基化反应电泳图
2.1.2 荧光光谱分析
荧光光谱技术的应用能够较为灵敏的反应蛋白质三级结构的变化。如图2所示,大豆11S蛋白与不同分子质量葡聚糖发生糖基化反应后生成轭合物的最大荧光强度值与未处理的大豆11S蛋白相比发生了不同程度的下降,说明糖基化反应影响了蛋白质的三级结构。葡聚糖多糖链与蛋白质的色氨酸残基结合后,对色氨酸残基产生屏蔽作用,其被包围在疏水环境中使蛋白具有更加紧密的三级结构[12],从而导致轭合物最大荧光强度的不断下降。如图2a和图2b,大豆11S蛋白与4、10 ku葡聚糖反应后最大荧光强度值在4 h最低;如图2c,大豆11S蛋白与70 ku葡聚糖反应后最大荧光强度值在3 h最低,这说明反应3~4 h时对色氨酸的屏蔽作用最强。但在糖基化反应后期4~5 h时,最大荧光强度却出现上升状态,这可能是因为葡聚糖与大豆11S蛋白的进一步紧密结合,蛋白质色氨酸残基之间相互接近其芳香族部位通过疏水相互作用形成复合物,这种复合物会导致最大荧光强度的峰值再次上升[20]。荧光光谱图中最大荧光强度的变化证明了大豆11S蛋白发生了糖基化反应且该反应对蛋白结构造成一定影响。



图2 大豆11S蛋白分别与4、7、10 ku 葡聚糖发生糖基化反应后荧光强度变化
2.2 大豆11S蛋白湿法糖基化反应后蛋白结构分析
2.2.1 表面疏水性分析
蛋白质表面疏水性的强弱与蛋白质表面的疏水基团的变化情况密切相关,通过分析表面疏水性变化可以反映糖基化反应后蛋白结构的变化[21]。如图3,改性后的大豆11S蛋白-葡聚糖轭合物表面疏水性低于改性前,并随着反应时间的延长呈现出先增加后降低的轻微波动趋势。该波动趋势可能受两方面因素影响,一方面由于蛋白质与葡聚糖结合后,葡聚糖相应的糖链部分可能会包裹或覆盖位于蛋白质外层疏水性基团,多糖链的遮蔽作用使荧光探针难以与蛋白质分子内部的疏水基团结合[12],从而使得疏水性下降;另一方面随着糖基化反应程度加深,和蛋白质结合的葡聚糖也会携带部分疏水基团,并随着糖基化反应迁移到蛋白表面[22],从而提高疏水性。两方面因素的共同作用使得糖基化反应后疏水性发生不同程度的改变,但是总体呈现降低趋势。通过比较不同分子质量轭合物表面疏水性变化趋势及峰值点,低分子质量葡聚糖(4、10 ku)糖基化反应程度最大,表面疏水性下降最明显。

注:不同大小写字母,标“*”字母分别表示大豆11S蛋白~4 ku葡聚糖轭合物、大豆11S蛋白~10 ku葡聚糖轭合物、大豆11S蛋白~70 ku葡聚糖轭合物之间的显著性差异(P<0.05)。余同。图3 大豆11S蛋白糖基化反应后表面疏水性变化
2.2.2 Zeta电位分析
Zeta电位是表征溶液胶体稳定性和蛋白质带电荷情况的重要参数。如图4,在中性条件下,大豆11S蛋白和大豆11S蛋白-葡聚糖轭合物Zeta电位值均为负值,且轭合物Zeta电位绝对值大部分低于改性前,大豆11S蛋白表面有带负电荷的羧基基团,葡聚糖不带电荷,二者结合后仍带负电。同时,葡聚糖亲水性糖链与大豆11S蛋白发生共价结合后,对蛋白质表面电荷产生屏蔽作用[23],导致Zeta电位绝对值下降。溶液中单个轭合物分子和可溶性的蛋白大粒径聚集物同时存在,可能是Zeta电位呈现出波动变化的原因[24]。Zeta电位与表面疏水性变化趋势相似,与齐宝坤等[14]的研究结果一致。轭合物Zeta电位绝对值下降可能受到蛋白质和多糖种类的影响[25],大豆11S蛋白含有较多的含硫氨基酸,凝胶性好且结构不易被破坏[26];葡聚糖的存在能够更好地保护蛋白结构,使得蛋白处于一种相对紧凑的状态[27],两方面因素可能导致蛋白表面带电基团被遮蔽,内部带电基团不易暴露,Zeta电位下降。

图4 大豆11S蛋白糖基化反应后Zata电位变化
2.3 大豆11S蛋白湿法糖基化反应后理化性质分析
2.3.1 不同pH条件下溶解性分析
图5为反应4 h后溶解度的变化,其余反应时间的变化与之相似。在不同的反应时间条件下,在pH 3~9的范围内,大豆11S蛋白-葡聚糖轭合物的溶解度随着pH的增加表现出先降低后增加的趋势,在pH=5时,溶解度最小。在所有反应时间下,大豆11S蛋白与不同分子质量的葡聚糖结合后生成的轭合物的溶解度均表现出不同程度的降低,且随着分子质量增大,溶解度降低程度增加,这可能是由于糖基化反应程度加深,亲水性糖残基附着引起蛋白形成更加致密的结构,蛋白质内部基团不易暴露[28],溶解度降低。但在等电点附近(pH=5~6),大豆11S蛋白-葡聚糖(4、10 ku)轭合物溶解度高于未反应大豆11S蛋白,这可能是由于大豆11S蛋白在等电点附近因疏水相互作用而形成结构紧密的聚集体,阻碍了葡聚糖的进一步结合,同时已经结合在蛋白表面的多糖产生位阻效应以防止大豆11S蛋白进一步聚集而提高溶解性[29]。Xu等[30]也发现葡聚糖与肌原纤维蛋白湿法糖基化反应后轭合物在等电点附近溶解性显著提高。在非等电点环境中,大豆11S蛋白结构容易受到pH的影响发生破坏,蛋白结构展开,体现出较好的溶解性,对其进行糖基化反应反而会生成不溶性大分子产生不利影响。低分子质量的葡聚糖形成的轭合物在碱性条件下比高分子质量葡聚糖形成的轭合物具有更高的溶解性,这可能是由于低分子质量多糖的空间位阻更小,更容易靠近蛋白质的氨基,糖基化反应的程度更大[31]。
大豆11S蛋白结构紧密,粒径分布大于100 nm,溶解性较差[9];葡聚糖具有良好的水溶性,但其糖基化修饰改善溶解度的效果不如葡萄糖、乳糖[32]。因此在对大豆11S蛋白进行改性的过程中,应选择能够提供更多羟基基团的糖,同时辅以一些其他手段(如:酶解、超声等)进一步破坏大豆11S蛋白的结构,对溶解性的改善会更加明显。

图5 大豆11S蛋白糖基化反应4 h后在不同pH 条件下溶解度变化
2.3.2 乳化活性(EA)及乳化稳定性(ES)分析
如图6所示,改性后的大豆11S蛋白-葡聚糖轭合物EA明显低于改性前,EA随反应时间的变化趋势不明显,这可能是由于糖基化反应引入了更多的亲水性糖残基,破坏了水油界面平衡[33],导致EA下降。大豆11S蛋白-4 ku葡聚糖轭合物EA最低,大豆11S蛋白-10 ku葡聚糖轭合物EA最高,这可能是受引入亲水糖链和糖链所带来的空间位阻的影响,低分子质量葡聚糖引入糖链多,降低EA;高分子质量葡聚糖糖链产生空间位阻大,提高EA。Dickinson等[34]和江连洲等[35]发现EA与表面疏水性、Zeta电位呈正相关,与本文结果相似。
改性后的大豆11S蛋白-葡聚糖轭合物ES明显高于改性前,ES随反应时间延长呈现出先增加后减小的趋势,3种轭合物都在反应4 h时,ES达到最大值。EA降低,反而导致ES上升,这意味着除了糖基化所带来的对油水界面破坏外,ES更多受放置后油水界面形成的保护膜稳定性影响,随着糖基化反应时间延长轭合物在形成乳状液的过程中与油滴的起始界面相应变小,但接触面一旦形成后,在随后放置的过程中油水界面形成的保护膜反而更加稳定[36],ES上升。而当反应进行到后期时,所形成的产物(褐变产物)会导致溶液絮凝和聚集,对ES产生了不利的影响[37]。
因此,不同分子质量葡聚糖糖基化反应改性大豆11S蛋白后对EA的影响是不利的,但对ES的影响是有利的,为了获得更好的ES,低分子质量(4 ku)葡聚糖、反应时间4 h是理想的改性条件。


图6 大豆11S蛋白糖基化反应后乳化性和乳化稳定性变化
2.3.3 持水性(WRC)及持油性(ORC)分析
如图7,改性后的大豆11S蛋白-葡聚糖轭合物WRC均上升,ORC均下降。这可能是因为湿法糖基化反应更加充分,增大了蛋白质、糖与水分子的接触面积,由于糖基化反应中大豆11S蛋白的表面疏水性基团可能与葡聚糖糖链结合,使其丧失在原有体系中相关疏水性的表达,疏水基团不易暴露,因此WRC上升,ORC下降[38,39]。但随着糖基化反应的不断进行,对蛋白结构的破坏也使得疏水基团暴露,因此WRC也会出现下降趋势,ORC出现上升趋势。两方面因素的共同作用导致WRC和ORC发生不断地波动,这与表面疏水性的波动变化一致。大豆11S蛋白-70 ku葡聚糖轭合物由于参与反应葡聚糖分子质量较大,糖基化反应速率较慢,WRC和ORC波动并不明显。总体而言,湿法糖基化反应能够有效改善大豆11S蛋白的WRC,抑制ORC,低分子质量(4和10 ku)的葡聚糖对WRC的提高作用和对持油性的降低作用更明显。
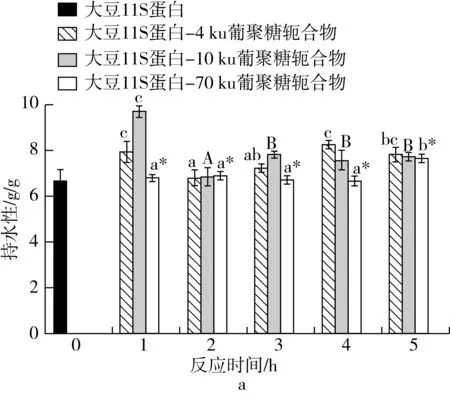

图7 大豆11S蛋白糖基化反应后持水性和持油性变化
3 结论
大豆11S蛋白与不同分子质量(4、10、70 ku)的葡聚糖在湿法条件下发生了糖基化反应,通过SDS-PAGE和荧光光谱分析证实了大豆11S蛋白-葡聚糖轭合物的形成,同时也表明糖基化反应程度随时间延长逐渐加深,4 h是理想的改性时间。糖基化反应过程中亲水性糖链的附着使得大豆11S蛋白结构更加致密,破坏水油界面平衡,内部基团不易暴露,因此大豆11S蛋白-葡聚糖轭合物与未处理的大豆11S蛋白相比表面疏水性、Zeta电位值、ORC、EA和在非等电点处(pH=3.0~4.0,7.0~9.0)溶解性均下降,WRC上升;而在等电点附近(pH=5.0~6.0)大豆11S蛋白易形成聚集物会阻碍葡聚糖进一步结合,同时已结合的葡聚糖产生空间位阻效应使得轭合物溶解性增加,ES的上升主要与放置后油水界面形成的保护膜的稳定性有关。另外,低分子质量葡聚糖对大豆11S蛋白湿法糖基化改性程度最大,对功能性质的影响更明显。由于大豆11S蛋白结构紧密,糖基化修饰对增强大豆11S蛋白表面活性的效果有限,为了获得更加理想的表面活性,选择一些辅助手段对大豆11S蛋白结构进行破坏、暴露更多内部基团以获得更好的糖基化效果是未来研究趋势。
