新疆维汉两民族早期结直肠癌患者肠道菌群的分布情况研究
2021-02-03贾腾飞
贾腾飞,王 理
1.新疆医科大学第一附属医院胃肠肿瘤外科,新疆 乌鲁木齐 830000; 2.浙江大学医学院附属第二医院急诊医学科
人类肠道中存在大量微生物,有证据表明,肠道菌群失调与多种疾病的发展及预后有关,尤其是结直肠癌(colorectal cancer,CRC)[1]。流行病学研究表明,长期食用富含饱和脂肪的红肉和加工肉会增加肠内黏膜摄入杂环胺,N-亚硝基化合物(NOC)等致癌物质,增加患结肠癌风险[2],由于新疆维汉两民族之间的饮食习惯不同,肠道菌群也不完全相同,研究显示,在维吾尔族结肠癌患者中优势菌乳酸杆菌量甚少,拟杆菌属、梭杆菌属数量较多[3]。因此,本研究拟分析维汉两民族之间早期CRC患者肠道菌群的分布情况,为后续临床研究提供研究基础。
1 资料与方法
1.1 一般资料选取2019年3月至2020年3月于新疆医科大学第一附属医院首次手术并经病理学证实为早期CRC的患者83例。根据不同民族分为汉族组和维吾尔族组,其中汉族组43例,男35例,年龄(58.25±4.35)岁;维吾尔族组40例,男31例,年龄(56.53±3.25)岁。两组性别、年龄、体质量指数等一般资料比较,差异无统计学意义(P>0.05),具有可比性(见表1)。
纳入标准:(1)术后病理分期符合2017年国际抗癌联盟/美国癌症联合委员会(UICC/AJCC)TNM 分期中的Ⅰ期的CRC[4];(2)东部肿瘤合作组(ECOG)的功能状态为0或1[5],其他器官功能正常;(3)年龄≥18岁。
排除标准:(1)手术治疗前曾经接受过胃肠道手术或放化疗治疗;(2)影像学检查资料显示存在其他部位肿瘤;(3)合并其他类型的肠道系统疾病;(4)合并其他感染性疾病。

表1 两组患者一般临床资料比较Tab 1 Comparison of general clinical data between the two groups
1.2 方法
1.2.1 粪便细菌常规培养:收集两组患者术前5 d内的新鲜粪便标本,将0.1 g粪便标本与0.9 ml 0.9%氯化钠注射液加入一次性离心管中进行混匀,厌氧菌37.5 ℃培养24~48 h,需氧菌37 ℃培养18~24 h。使用平板计数法对培养菌株计数和鉴定。按照下列公式计算细菌集落数:
克粪便活菌集落数(log10)=(标本质量+稀释量)/稀释度×标本质量×菌落个数×(稀释度和稀释倍数)。
通过菌落形态和纯度,以及革兰染色结果对细菌种类进行鉴定。
1.2.2 肠道菌群的高通量测序:取0.5 g待测粪便标本,DNA缓冲液稀释后,800 r/min离心5 min,离心半径6 cm,使用QIAamp DNA Stool Mini Kit试剂盒(德国Qiagen公司)提取粪便标本细菌总DNA,测定浓度后稀释为40 ng/μl,放入-80 ℃中保存。采用Illumina Miseq高通量测序平台在16S rDNA V3~V6区进行PCR扩增,鉴定细菌的聚类成操作分类单元(OTU),使用Mothur软件计算多样性指数测序深度指数(%)和菌群多样性指数,寻找丰度>50%的序列进行物种分类,评估各肠道菌群的结构丰度。
1.2.3 粪便细菌内肠道菌群的变化:根据高通量测序结果将待测的粪便标本DNA进行大肠埃希氏菌、具核梭杆菌、梭状芽胞杆菌、脆弱拟杆菌4种细菌的实时定量荧光PCR反应,具核梭杆菌PCR试剂盒选用Prime Script RT-PCR试剂盒(日本TAKARA),其余试剂盒均购自北京百奥莱博公司。细菌内参基因16S rDNA引物序列:上游引物为5′-CCATGAAGTCGGAATCGCTAG-3′;下游引物为5′-GCTTGACGGGCGGTGT-3′。SYBR mix 10 μl,总DNA 2 μl,ddH2O 7 μl,目标细菌上下游引物各1 μl。反应条件:95 ℃预变性2 min,95 ℃变性30 s,55 ℃退火30 s,74 ℃延伸30 s,循环45次,采用微量核酸分析仪(德国IMPLEN公司)计算基因相对表达。
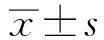
2 结果
2.1 高通量测序分析结果结果显示,OUT数量随着测序量的增加而增加,Shannon-Wiener曲线逐渐平坦,Shannon指数也逐渐饱和,表明测序深度合理,测序数据量大。共鉴定出10个细菌门类和110个细菌属类。两组均以厚壁菌门、拟杆菌门、变形菌门、梭杆菌门的丰度较高。在属分类上而两组丰度较高的细菌属类有肠杆菌属、梭状芽胞杆菌属、拟杆菌属、梭杆菌属(见表2)。
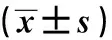
表2 肠道菌群多样性比较Tab 2 Comparison of intestinal flora diversity
2.2 肠道菌群情况对比分析粪便细菌常规培养结果显示,肠杆菌、肠球菌、双歧杆菌、乳酸菌在两组之间比较,差异均无统计学意义(P>0.05)(见表3)。高通量测序结果显示,两组均以厚壁菌门、拟杆菌门、变形菌门、梭杆菌门的丰度较高,且两组之间各细菌门类比较,差异均无统计学意义(P>0.05)。在细菌属水平上,两组之间比较,差异无统计学意义(P>0.05)(见表4)。两组大肠埃希氏菌、具核梭杆菌、梭状芽胞杆菌、拟杆菌属的基因相对表达比较,差异均无统计学意义(P>0.05)(见表5)。

表3 粪便细菌培养结果比较Tab 3 Comparison of fecal bacterial culture results

表4 肠道细菌组分的比较Tab 4 Comparison of intestinal bacterial components %)
表5 肠道特殊菌群的基因相对表达量比较Tab 5 Comparison of gene relative expression of intestinal special flora
3 讨论
人类肠道微生物组在调节人类体内稳态和代谢中的作用和贡献而被公认为“微生物器官”。研究显示,肠道微生物组的变化可以介导或改变环境因素对患CRC风险的影响[6]。影响患CRC风险的因素也影响肠道微生物组,这些因素会改变微生物组的结构和功能,以及介导CRC的代谢和免疫途径[7]。因此,有学者认为可以将肠道微生物组作为CRC早期预防策略的一部分。
本研究结果显示,维汉两民族早期CRC患者常见的粪便细菌中以肠杆菌、肠球菌、双歧杆菌、乳酸菌多见,且两组之间差异无统计学意义,既往研究[7-8]已经证实大肠埃希菌为主的大肠杆菌和肠球菌均为条件致病菌,当体内大量存在时会对机体造成不同程度损伤。双歧杆菌和乳酸杆菌是益生菌,有研究[8]显示,对于肠道手术患者早期给予双歧杆菌等益生菌可以有效降低术后机体应激反应。
本研究通过高通量测序检测了维汉两民族早期CRC患者中的肠道菌群,结果显示,在细菌门水平上,主要由厚壁菌门、拟杆菌门、变形菌门、梭杆菌门组成。在菌属水平上,以拟杆菌属占比较大,其次为肠杆菌属、梭菌属、梭杆菌属,但均未超过5%。多项研究已经证实,与健康个体相比,脆弱拟杆菌、大肠埃希氏菌、梭状芽胞杆菌、具核梭杆菌分别作为拟杆菌属、肠杆菌属、梭菌属、梭杆菌属的代表性细菌,在结直肠腺瘤或腺癌患者中的均大量存在[9-10]。并且在维吾尔族结肠癌患者中拟杆菌属、梭杆菌属数量较多[3]。这些肠道细菌通过各种机制参与CRC的发生和发展,如致癌物质的产生、炎症的诱导、宿主信号传导、免疫系统的调节等[11]。
其中肠毒素性脆弱拟杆菌(ETBF)已经被认为是CRC的一个最重要的危险因素,血清学检测抗BFT抗体可能有助于检测ETBF水平的升高,从而及早检测亚临床结肠损伤[12]。但是产生脆弱类杆菌毒素的ETBF对结肠上皮细胞的黏附及其对致癌作用的影响尚待证实,尽管有大量的研究证据表明ETBF与CRC之间存在关联,但流行病学研究的证据很少[6,13]。并且不同研究之间的ETBF阳性CRC的比例不同(26%vs89%),可能是由于分析方法或样品处理方法存在差异。此外,作为肠道菌群中最为常见的大肠埃希氏菌又称大肠杆菌,是变形菌门中常见的细菌之一,近期一项来自荷兰的研究进一步证实了大肠埃希氏菌携带的一种保守的致病性基因“聚酮合成酶(polyketide synthase,pks)”岛,可产生小分子毒性物质colibactin,导致肠道内膜细胞双链内交联和独特的DNA损伤模式,导致结肠上皮紧密连接的丧失,从而增加了大肠杆菌对肿瘤组织的内化作用[14]。本研究中发现大肠埃希菌数量较小,主要是因为该细菌多定殖在结肠黏膜上并进入肠细胞内,在粪便细胞中的残留较少[15]。
本研究还发现,在维汉两民族早期CRC患者中肠道细菌中还有一定数量的梭状芽胞杆菌和具核梭杆菌,多项队列研究证实与非肿瘤标本相比,CRC患者中梭状芽胞杆菌DNA和RNA序列的水平增加[16-17]。该细菌主要通过产生脱氧胆酸(DCA)导致胃肠道毒性[16],DCA是由特定种类的肠道细菌产生的次级胆汁酸。通过产生活性氧和氮,从而导致DNA损伤并增强对凋亡的抵抗力[17]。一项来自日本的队列研究代谢组学分析显示,患有多息肉样腺瘤(CRC的前体)的患者粪便中DCA的浓度较高[1]。针对具核梭杆菌,研究已经证实从癌前病变到CRC具核梭杆菌rDNA的拷贝数和相对百分比逐渐升高,显著高于未发生组织学改变的正常黏膜组织[18]。并且发现较高的具核梭杆菌的含量与疾病的晚期阶段,较高的复发风险和较短的患者生存时间有关[19-20]。流行病学研究显示,具核梭杆菌具有CRC锯齿状瘤形成的特定临床和分子特征,如BRAF突变和微卫星不稳定的过度突变[19]。具核梭杆菌在粪便中的富集,也同样被认为是从结肠腺瘤发展到癌症的潜在危险因素[20]。
由于维吾尔族患者与汉族患者在饮食习惯上的长期差异,对于红肉或加工肉的摄入比例不同,国际癌症研究机构世界卫生组织癌症研究机构的研究认为红肉和加工肉摄入量增加与CRC发病率增加有关[21]。荟萃分析表明,每日摄入100 g红肉和加工肉,患CRC的风险增加12%,主要致癌因素包括血红素化合物,杂环胺,NOC和未消化的蛋白质等[22]。这些分子除了具有直接的致癌作用外,还可以修饰肠道菌群,从而影响基因表达和结直肠上皮细胞稳态,从而促进CRC的发展[2]。长期摄入猪肉等红肉,而不是白肉或非肉类,血浆和尿液中肉碱和氧化三甲胺(TMAO)的水平升高[23]。其中肉碱在脂肪酸代谢中发挥重要作用,血液中高浓度的肉碱可作为代谢功能失调的潜在生物标志物[24]。而胆碱-TMAO途径也被证明在CRC的发展中具有潜在作用[9]。并且大量红肉也会增加次级胆汁酸的产生,研究均证明CRC相关微生物与脂肪和肉类饮食之间存在代谢联系[25]。但本研究结果显示,早期CRC患者中维汉两民族之间的肠道菌群比例差异无统计学意义。我们认为一方面是因为本研究样本量较小,而且新疆地区的高脂肪饮食特性,仍需大量数据证实该结论,另一方面尽管饮食成分或其微生物代谢是肠道菌群功能和组成的重要决定因素。微生物营养不良可以诱导宿主基因表达和炎症反应改变,从而促进致癌的微环境。但是饮食与癌症关联背后的复杂代谢和炎症机制仍不清楚[6-7]。
本研究局限性在于,仅评估了早期CRC患者中肠道菌群的分布情况,未能对肠道微生物菌群之间的相互作用机制做进一步研究,由于肠道菌群非常复杂,从单个微生物、宏基因组或代谢数据集中预测CRC会导致准确性降低,还需要考虑不同其他微生物物种之间以及微生物与宿主之间的相互作用。其次,还需要进一步多中心大样本量研究证实不同肠道菌群的分布对CRC患者术后并发症的影响,如术后感染及肠梗阻等。
总之,本研究结果显示,在早期CRC患者中,拟杆菌属含量较高,其次为梭菌属、梭杆菌属、肠杆菌属,并且在维汉之间差异无统计学意义。因此,微生物组的变化可用作早期检测CRC的生物标记,以改善筛选策略。
