GC法测定三香散中桂皮醛、反式茴香脑含量
2021-01-30陈骁勇葛玉松支荣荣
陈骁勇 葛玉松 支荣荣▲
1.江苏省盐城市第一人民医院药剂科,江苏盐城 224000;2.江苏省盐城市食品药品监督检验中心,江苏盐城 224000
三香散是盐城市中医院的自制制剂,由木香、小茴香、肉桂三味药构成,具有温中散寒、除胀止痛的功效,临床上常用于小儿肠绞痛以及寒泻而致腹胀、腹痛,为儿科腹痛腹胀的外用制剂[1-3]。目前,三香散标准为《江苏省食品药品监督管理局医疗机构制剂标准》JSZBZ20091017Z[4],标准中只有显微、理化鉴别等项目,缺乏定量测定的内容,其中肉桂、小茴香为三香散的主要成份[5-8]。本研究参照有关文献[9-15],建立气相色谱法(GC)同时测定三香散中桂皮醛和反式茴香脑含量的方法,旨在为该医院制剂三香散提供更好的标准和更新的依据,现报道如下。
1 仪器与材料
1.1 仪器
安捷伦7890A GC System 气相色谱仪(Agilent Technologies),XS205 DU 电子天平(Mettler Toledo);QLB型纯净空气泵(山东塞克赛斯氢能源有限公司);SPH-300 氢气发生器(北京中惠普分析技术研究所);A10 TOC monitor 实验室纯水(默克化工技术有限公司)
1.2 药品与试剂
1.2.1 药品 三香散(盐城市中医院自制制剂,规格:100 g,批号:191102、191120、191218);肉桂、小茴香、木香均来自盐城市第一人民医院药剂科;桂皮醛对照品(批号:110710-201619,含量:98.9%)、反式茴香脑对照品(批号:111835-201804,含量:99.6%)均购自中国食品药品检定研究院。
1.2.2 试剂 超纯水;乙酸乙酯为分析纯。
2 方法与结果
2.1 色谱条件
色谱柱:Agilent DB-WAX 色谱柱(30 m×0.320 mm,0.25 μm);柱程序升温:初始110℃,保持2 min,5℃/min升温至160℃,运行至12 min,30℃/min 升温至220℃,保持5 min,50℃/min 降温至110℃,保持2 min;进样口温度:250℃;FID 检测器温度:300℃;载气:高纯H2(体积分数>99.999%);流速:30 mL/min;进样量:1 μL;分流比:10∶1。
2.2 溶液的制备
2.2.1 混合对照品溶液 分别精密称定桂皮醛对照品、反式茴香脑对照品430.33 mg 和46.12 mg,分别置于10 mL容量瓶中,加乙酸乙酯适量使容量瓶中对照品溶解,并稀释至刻度,摇匀,制成质量浓度分别为43.033 mg/mL、4.612 mg/mL 的桂皮醛对照品储备液和反式茴香脑对照品储备液;精密量取上述桂皮醛对照品储备液0.1 mL 与反式茴香脑对照品储备液1.5 mL,分别置于25 mL 容量瓶中,加乙酸乙酯稀释至刻度,摇匀,制成浓度分别为0.1721 mg/mL 和0.2767 mg/mL的混合对照品溶液。
2.2.2 供试品溶液 取三香散供试品,混合均匀,精密称取0.5000 g,置于250 mL 具塞圆底烧瓶中,加入适量超纯水与2 mL 乙酸乙酯,连接挥发油测定器,缓缓加热至沸,并保持微沸约5 h,放冷至室温,取上层乙酸乙酯液至5 mL 容量瓶中,用乙酸乙酯定容至刻度,摇匀,用0.45 μm 有机微孔滤膜滤过,取续滤液,即得供试品溶液。
2.2.3 阴性样品溶液 按照三香散处方中各饮片成分比例和该制剂的制备工艺,称取0.2 g 木香、0.2 g 小茴香,制成不含肉桂的阴性样品;称取0.2 g 木香、0.1 g肉桂,制成不含小茴香的阴性样品。按照“2.2.2”项下流程,制备不含有相应饮片的阴性样品溶液,即缺小茴香的阴性样品溶液,缺肉桂的阴性样品溶液。
2.3 线性关系考察
精密量取“2.2.1”项下的桂皮醛对照品储备液1 mL,置于20 mL 容量瓶中,用乙酸乙酯稀释至刻度,分别取0.025、0.050、0.100、0.200、0.500 mL 置于25 mL容量瓶中,加乙酸乙酯稀释至刻度,用0.45 μm 有机微孔滤膜滤过,以1 μL 自动进样至气相色谱仪,测定各峰面积。以桂皮醛对照品浓度(X1)为横坐标,桂皮醛对照品峰面积为纵坐标(Y1),绘制出标准曲线,得到桂皮醛的回归方程为Y1=1948X1+36.908(R2=0.9995),桂皮醛在浓度0.00213~0.042 56 mg/mL 范围中呈现良好的线性关系(图1A)。
精密量取“2.2.1”项下反式茴香脑对照品储备液0.4、0.8、1.0、1.5、5.0 mL,分别置于25 mL 容量瓶中,用乙酸乙酯稀释至刻度,同样用0.45 μm 有机微孔滤膜滤过,以1 μL 自动进样至气相色谱仪,测定各峰面积。以反式茴香脑对照品浓度(X2)为横坐标,反式茴香脑对照品峰面积为纵坐标(Y2),绘制出标准曲线,得到反式茴香脑的回归方程为Y2=3425.3X2-62.95(R2=0.9996),反式茴香脑在浓度0.0734~0.9180 mg/mL范围中呈现良好的线性关系(图1B)。
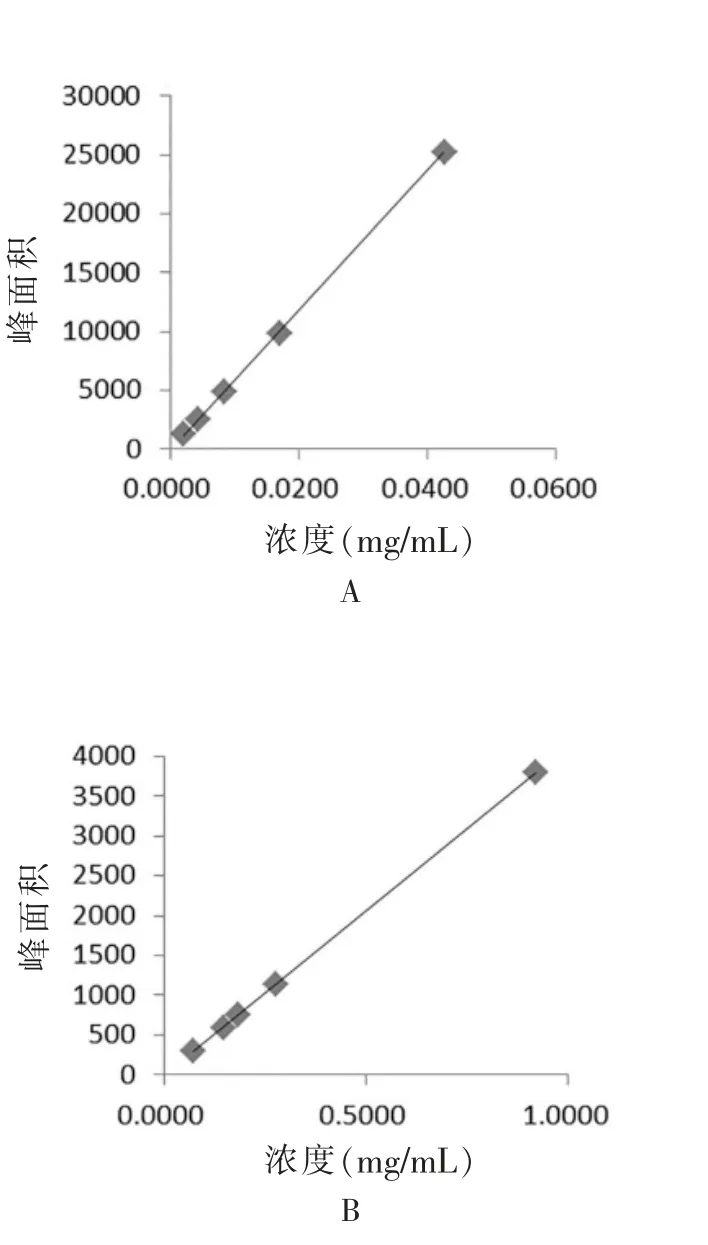
图1 标准曲线图
2.4 方法学考察
2.4.1 精密度试验 按照“2.1”项下色谱条件,以1 μL自动进样“2.2.1”项下制备的混合对照品溶液6次,至气相色谱仪,记录6次峰面积。计算得出,桂皮醛、反式茴香脑峰面积的RSD 分别为1.50%、0.84%,结果表明仪器精密度良好。
2.4.2 专属性实验 分别取“2.2”项下的供试品溶液、混合对照品溶液、缺小茴香阴性样品溶液,缺肉桂阴性样品溶液,按照“2.1”项下色谱条件进行测定。结果显示,主成分峰与杂质峰之间分离度达到要求,原辅料均无干扰(图2)。
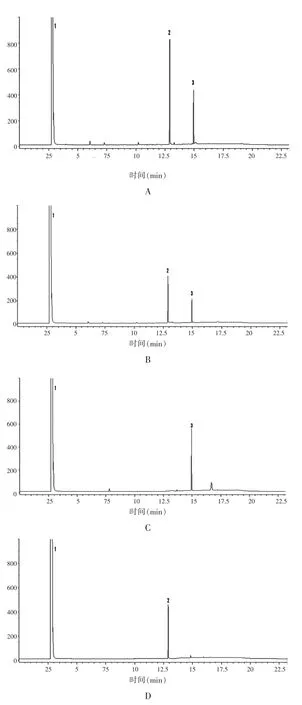
图2 专属性试验气相色谱图
2.4.3 稳定性试验 精密称取三香散粉末(批号:191218)按照“2.2.2”项下方法制备供试品溶液,分别置于室温下0、1、2、4、8、12、24 h,按照“2.1”项的色谱条件自动进样1 μL 于气相色谱仪,分析测得桂皮醛、反式茴香脑的RSD值分别为1.44%、0.84%。结果表明,桂皮醛、反式茴香脑供试品溶液中,成分桂皮醛、反式茴香脑在室温下24 h 内稳定。
2.4.4 重复性试验 精密称取三香散粉末(批号:191218)按照“2.2.2”项下方法制备供试品溶液,按照“2.1”项的色谱条件自动进样1 μL 于气相色谱仪进行进样分析,得到桂皮醛、反式茴香脑的平均含量分别为0.1625、0.2473 mg/g,RSD值分别为0.96%、0.48%,结果表明,本实验方法的重复性良好。
2.4.5 加样回收率实验 精密称取三香散粉末(批号:191218)6份,分别加入质量浓度为0.1721 mg/mL 和0.2767 mg/mL 的混合对照品溶液1 mL,精密加入2 mL乙酸乙酯,加入适量超纯水,按照“2.2.2”项下方法制备供试品溶液,按照“2.1”项下色谱条件自动进样1 μL于气相色谱仪进行进样分析,计算回收率,结果见表1。结果显示,两种成分的平均回收率均>9.9%,RSD≥1.50。
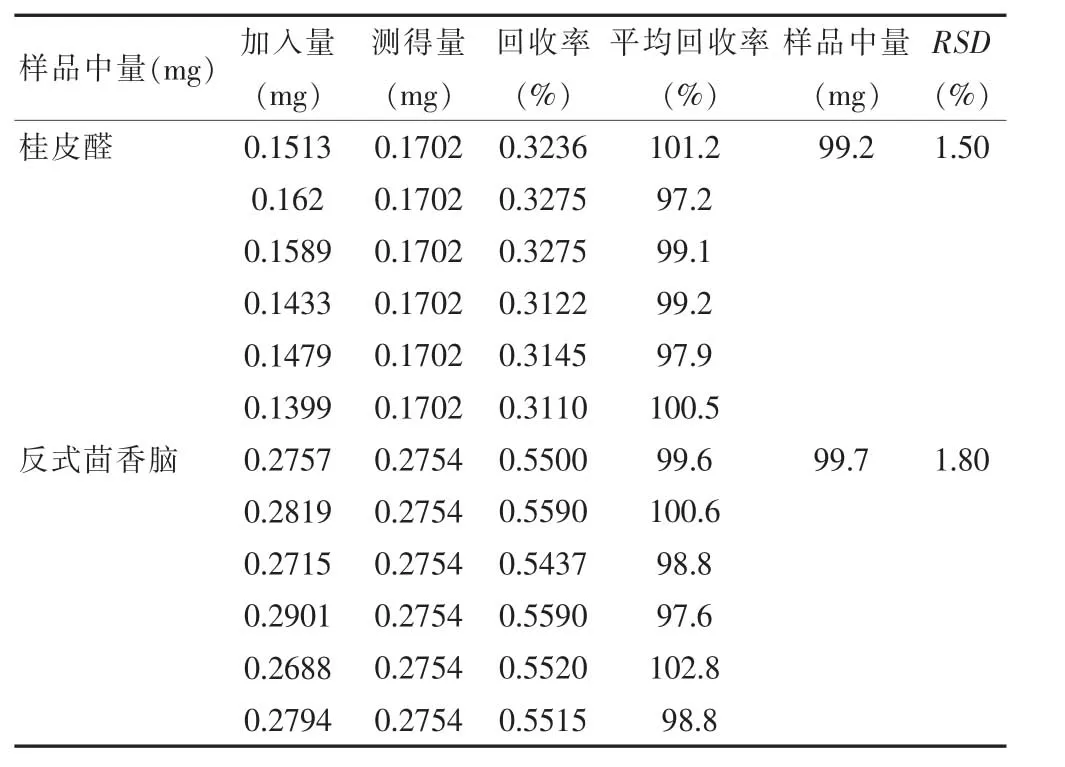
表1 加样回收率实验测试结果
2.4.6 样品含量测定 取3个不同批号的三香散制剂(批号:191102、191120、191218),按照“2.2.2”项下方法制备供试品溶液,分别吸取3 批次供试品溶液各6次,按照“2.1” 项下色谱条件自动进样1 μL 于气相色谱仪进行进样分析,记录各峰面积,计算桂皮醛和反式茴香脑的含量,结果见表2。结果显示,批号191120的三香散中,桂皮醛和反式茴香脑的含量均最高,分别为0.1611 mg(RSD=1.10%)和0.2555 mg(RSD=1.20%)。
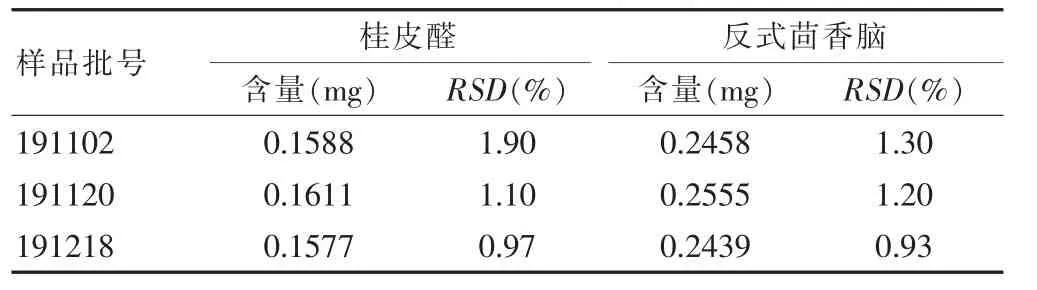
表2 样品含量测定(n=6)
3 讨论
3.1 色谱柱选择
分别选取Agilent DB-624 色谱柱(30 m×0.530 mm,3.00 μm)和Agilent DB-WAX 色谱柱(30 m×0.320 mm,0.25 μm),对样品中的两种化学成分进行分析。结果显示,采用Agilent DB-WAX 色谱柱(30 m×0.320 mm,0.25 μm),两种化学成分的分离度>1.5,理论塔板数较高,因此本研究选取Agilent DB-WAX 色谱柱。
3.2 成分提取
在成分提取过程中,采用超声提取20 min 与水蒸气蒸馏法1 h 进行对比,结果显示,水蒸气蒸馏法提取1 h,能将桂皮醛和反式茴香脑成分提取完全,并且操作简便易行。本研究选取水蒸气蒸馏法提取5 h,旨在使桂皮醛和反式茴香脑成分提取更加完全。
3.3 温度选择
由于三香散药味多芳香,在分离各成分时发现,恒定柱温无法有效分离各成分,经过比较沸点、初始温度、升温速率,末温等因素,确定了程序升温。结果显示,初始110℃,保持2 min,5℃/min 升温至160℃,运行至12 min,30℃/min 升温至220℃,保持5 min,50℃/min 降温至110℃,保持2 min,上述程序升温方法能够使低沸点组分和高沸点组分在色谱柱中有适宜地保留,各待测组分色谱峰分离度较好,且峰形比较对称。
3.4 小结
本实验采用了GC 测定三香散中桂皮醛、反式茴香脑的含量,方法学研究显示,实验中采取的方法能够有效地对三香散中的桂皮醛和反式茴香脑进行含量测定,具有方便简便、准确、省时、重复性好的特点,并且能够弥补现有鉴定标准中对三香散各成分的定量鉴别内容的空白,为制剂充分发挥其治疗小儿腹痛、腹胀、胃肠痉挛等症状的功效,提供理论依据。
