高效液相色谱法对食品中苯并芘含量测定的不确定度评定
2021-01-28刘德群周铭林何海彤
王 邱,刘德群,周铭林,江 琦,何海彤
(广东产品质量监督检验研究院,广东佛山 528000)
苯并芘又称苯并(a)芘、3,4-苯并芘,英文缩写为 Bap,是一种常见的高活性间接致癌物和突变原[1]。若被释放至大气中以后,容易与大气中各种类型的微粒所形成的气溶胶结合在一起,在8 μm以下可吸入尘粒中,易经呼吸道吸入至肺部,进入肺泡甚至血液,从而导致肺癌和心血管疾病[2-3]。
测量不确定度的评定工作是分析测试实验中一项非常重要的内容,它能够定量反映测量结果正确性的可疑程度[4]。一个完整的测量结果应至少含有两个基本量:一是被测量的最佳估计值;二是表述该测量结果的离散型程度即为测量结果不确定度[3,5-7]
本研究依据 JJF 1059—1999《测量不确定度评定与表示》[8-10]和JJF 1135—2005《化学分析测量不确定度评定》[11-13],按照GB 5009.27—2016《食品安全国家标准食品中苯并(a)芘的测定》[14]对三种不同基质试样(大米试样、烟熏肉试样、食用植物油试样)进行处理,分别对不同试样中苯并芘含量测定过程中的不确定度的各个分量进行评定,并给出扩展不确定度,以期建立一套合理、完整的评定方案,可以作为改善食品中苯并芘含量测定的测量程序,提高其测量准确性和精密度等方面的理论依据。
1 材料与方法
1.1 材料与试剂
苯并(a)芘标准品(批号为 H419128AL,浓度为10.00 mg/L):德国DR公司;大米试样:华润五丰米业(中国)有限公司;烟熏肉试样:佛山市顺德区东龙烤鳗有限公司;食用植物油试样:东莞鲁花食用油有限公司;苯并(a)芘分子印迹柱(货号为 50610020,规格为 200 mg,3 mL):上海安谱公司;正己烷、二氯甲烷(均为色谱纯):美国honeywell公司;乙腈(色谱纯):美国TEDIA公司。
1.2 仪器与设备
超高效液相色谱仪,配置荧光检测器(型号为 LC30-AD,工作站为 LabSolution):日本SHIMADZU公司;电子天平(感量为1 mg,0 g≤m≤50 g时,最大允许误差为0.005 g):广东省中科进出口有限公司。
1.3 方法
1.3.1 标准溶液的配制
吸取1.0 mL标准溶液于100 mL容量瓶中,用乙腈定容,得到100 ng/mL苯并(a)芘标准储备液。系列标准工作溶液配制方法见表 1。用乙腈配制成0.5、1、2、5、10、20 ng/mL 6个浓度点,每个浓度重复进样三次,以期获得峰面积与浓度之间的线性关系以考察标准曲线拟合所引入的不确定度。

表1 标准工作溶液的配制Table 1 Preparation of standard working solution
1.3.2 样品前处理方法
1.3.2.1 大米试样 称取1 g(精确到0.001 g)试样(每个试样称2次,取两次称量结果的平均值),加入5 mL正己烷,旋涡混合0.5 min,40 ℃下超声提取10 min,4 000 r/min离心5 min,转移出上清液。再加入 5 mL正己烷重复提取一次。合并上清液,转移至预先已活化完全的苯并芘分子印迹柱中(依次用5 mL二氯甲烷及5 mL正己烷活化柱子),将待净化液转移进柱中,待液面降至柱床时,用6 mL正己烷淋洗柱子,弃去流出液。用6 mL二氯甲烷洗脱并收集净化液到试管中。将净化液在40 ℃下氮气吹干,准确吸取1 mL乙腈涡旋复溶0.5 min,过微孔滤膜后供液相色谱测定。
1.3.2.2 烟熏肉试样 提取和净化方法同大米试样一致。
1.3.2.3 植物油试样 除称样0.4 g(精确到0.001 g)和最后用0.4 mL乙腈定容外,其余处理均与大米试样一致[14]。
1.3.2.4 空白试样 加标回收率实验前处理方法:按照上述前处理方法选出三种不同基质试样的空白样品(即含量为未检出),在空白大米试样、空白烟熏肉试样、空白食用油试样均添加 10 ng的标准溶液,每种试样各做6个平行,再按照上述前处理方法进行提取、过柱、氮吹以及复溶等步骤后过滤膜上机。
1.3.2.5 加标试样 选择其中一个加标试样重复上机测定6次以考察高效液相色谱仪重复进样引入的相对标准不确定度。
1.3.3 色谱条件
色谱柱:C18柱(250 mm×4.6 mm,4 μm);流动相:乙腈-水(88∶12,v/v)等度洗脱;流速:1.0 mL/min;激发波长384 nm,发射波长406 nm;柱温:室温;进样量:20 μL。
2 结果与分析
2.1 数学模型以及不确定来源分析
根据公式 X=C×V ×F×1 000/(m×1 000)
其中:X:试样中苯并(a)芘含量(μg/kg);C:从标准工作曲线得到被测组分的质量浓度(ng/mL);m:样品称样量(g);V:定容体积(mL);F:稀释因子,按照标准处理方法F=1;1 000:由ng/g换算成μg/kg的换算因子。
结果保留到小数点后一位。
从数学模型和实际实验过程来看,食品中苯并芘含量测定的不确定度主要来源于标准溶液配制、标准曲线拟合、样品前处理过程、高效液相色谱仪(重复测定)等方面。
2.2 不确定度分量的评定
2.2.1 标准溶液配制引入的不确定度
2.2.1.1 标准物质纯度引入的相对标准不确定度uref(标)经查苯并(a)芘标准品证书,标准溶液浓度为10.00 mg/L,其扩展不确定度为0.31 mg/L(k=2),按矩形分布处理[15],则标准溶液的相对标准不确定度为:
2.2.1.2 标准工作溶液配制过程中容量瓶引入的相对标准不确定度uref(容)和移液枪引入的相对标准不确定度uref(枪)标准工作溶液配制过程中容量瓶使用情况以及不同容量瓶的相对标准不确定度见表2。单标线10 mL容量瓶和单标线100 mL容量瓶经检定均为A级,服从三角分布[16],相对标准不确定度公式为。因此,标准工作溶液配制过程中容量瓶引入的总相对标准不确定度为

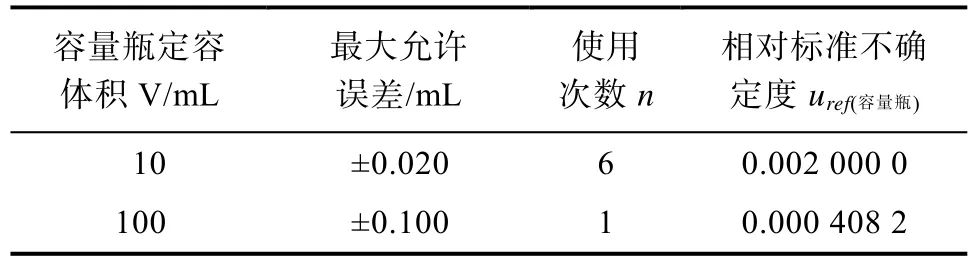
表2 标准工作溶液配制过程中容量瓶引入的不确定度Table 2 Uncertainties of volumetric flasks form calibration solution preparation
标准工作溶液配制过程中移液枪使用情况以及不同移液枪的相对标准不确定度见表 3。按照矩形分布计算[17],其相对标准不确定度公式为。因此标准工作溶液配制过程中移液枪引入的总相对标准不确定度为
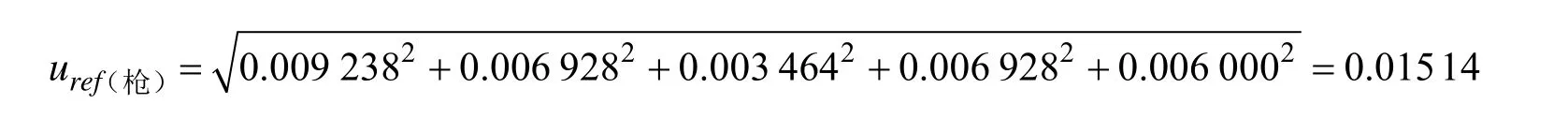

表3 标准工作溶液配制过程中移液枪的不确定度Table 3 Uncertainties of pipettes form calibration solution preparation
2.2.1.3 标准溶液配制过程中温度变化引起的溶剂体积改变引入的相对标准不确定度uref(溶剂)实验室温度在(20±3)℃范围内波动,玻璃器具受温度波动的影响较小,可忽略不计,但温度波动引起溶剂体积的改变需考虑。本次标准储备液和标准工作溶液均由乙腈配制,20 ℃时乙腈的体积膨胀系数(a)为1.37×10-3℃-1[18],相对标准不确定度公式为,ΔT=3 ℃,n为使用次数。标准溶液配制的全部过程均由容量瓶和移液枪完成,环境温度变化引入的容量瓶和移液枪中溶剂体积改变的相对标准不确定度见表4。 因此,环境温度变化引入的相对标准不确定度为:


表4 环境温度变化引入的不确定度Table 4 Uncertainties form temperature changes in the experimental environment
2.2.1.4 标准溶液配制过程的合成相对标准不确定度uref(1) 标准溶液配制过程引入的不确定度主要由标准物质纯度的不确定度、配制时容量瓶引入的不确定度、配制时移液枪引入的不确定度以及环境温度变化时乙腈体积改变引入的不确定度这几部分组成。

2.2.2 样品前处理过程引入的不确定度
2.2.2.1 称量试样时天平引入的不确定度uref(天平)样品前处理称样时使用的是千分之一天平,其最大允许误差为±0.005 g,属均匀分布[16],且样品称样量是由每个试样的两次称样所得,三种不同试样称量时天平引入的相对标准不确定度见表5。按照矩形公式计算,其相对标准不确定度公式为
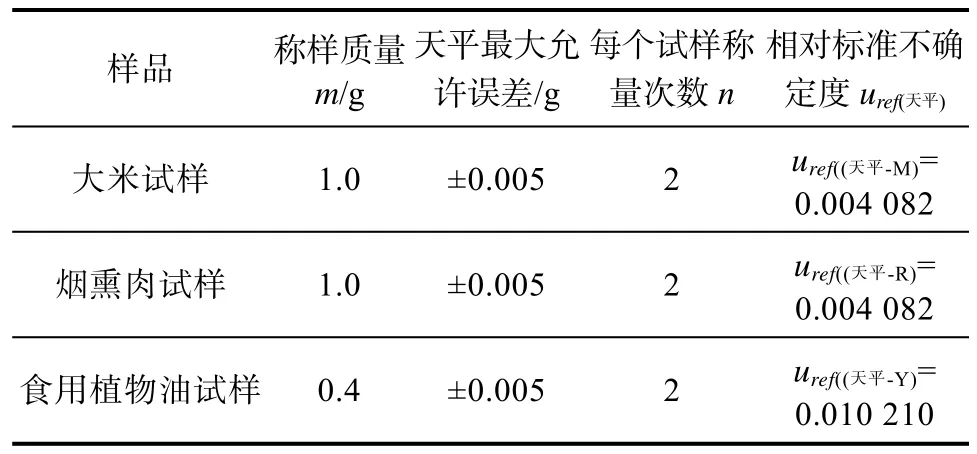
表5 三种不同试样称量时天平引入的不确定度Table 5 Uncertainties form weighing of electronic balance of three different samples
2.2.2.2 加标回收率引入的不确定度uref(回收)样品前处理过程较为复杂,需经过提取、均质、过柱净化、氮吹等步骤,难以对每个环节逐一进行评定,可通过对样品称样后加标测定其回收率的方式对样品前处理过程进行不确定度评定。不同样品加标回收率结果见表 6。加标回收率的平均值的标准不确定度。《测量不确定度表示指南》[19]中要求校正明显的系统误差,因此应进行t检验(公式为)以考察平均回收率R与100%之间的差异是否具有统计学意义,如果差异具有统计学意义(t≥t95,5=2.57),则需要用回收率校正因子f(f=1/)校正结果,应归入到样品质量分数的计算中,否则不必进行校正。三种不同基质试样加标回收率的相对标准不确定度和t检验结果见表7。

表6 三种不同基质试样6次平行试验的加标回收率Table 6 Recoveries of three different samples at six parallel experiments %
2.2.2.3 样品前处理过程引入的合成相对标准不确定度uref(2) 样品前处理过程引入的不确定度主要由称量试样时天平引入的不确定度和加标回收实验引入的不确定度两部分组成:uref(2)=。因此,三种不同基质试样前处理过程引入的合成相对标准不确定度为:

表7 三种不同基质试样加标回收率的不确定度和t检验结果Table 7 Uncertainties and t test results of recoveries of three different samples
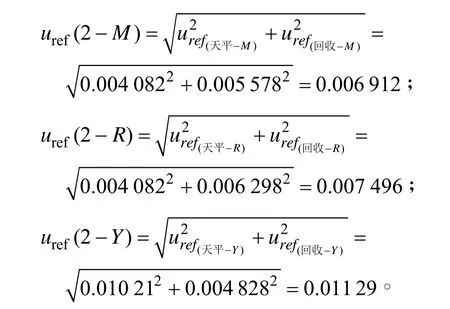
2.3.3 最小二乘法拟合标准曲线时引入的相对不确定度uref(3)
标准曲线拟合的不确定度由标准系列溶液进行测定得到。峰面积Y与浓度的线性关系表8。

表8 标准溶液的浓度-峰面积对应表Table 8 Concentration peak area of standard solutions
以三种不同基质的空白加标样品的峰面积经过线性回归方程计算后得到的质量浓度C为例,见表9。

表9 三种不同基质的空白加标样品的含量结果Table 9 Determine results of three different spiked samples ng/mL
标准曲线拟合时的不确定度公式[20]为:,其中S为标准曲线的标准偏差,公式为:b为线性回归方程的斜率,b=6 947.7,p为样品平行测定的次数,p=6;n为标准曲线每个浓度点测定的总次数,n=18;C0为样品的平均浓度;为所有标准溶液的质量浓度平均值,=6.4ng/mL;且三种不同基质试样的相对标准不确定度,见表10。

表10 三种不同基质试样的加标样品含量平均值的不确定度Table 10 Uncertainties of average content results of three different spiked samples
2.2.4 高效液相色谱仪重复进样引入的相对标准不确定度uref(4)


表11 加标试样6次重复进样每一针的浓度Table 11 Six concentrations of each injection of the spiked sample
2.3 合成相对标准不确定度Uref
综上所述,高效液相色谱法测定食品(大米类、烟熏肉类、食用植物油类)中苯并芘含量的相对标准不确定度由标准溶液配制过程引入的合成相对标准不确定度uref(1)、试样前处理过程引入的合成相对标准不确定度uref(2)、标准曲线拟合时引入的相对标准不确定度uref(3)和高效液相色谱仪重复进样引入的相对标准不确定度uref(4)这四个主要部分构成,不同基质试样的合成相对不确定度分别为:
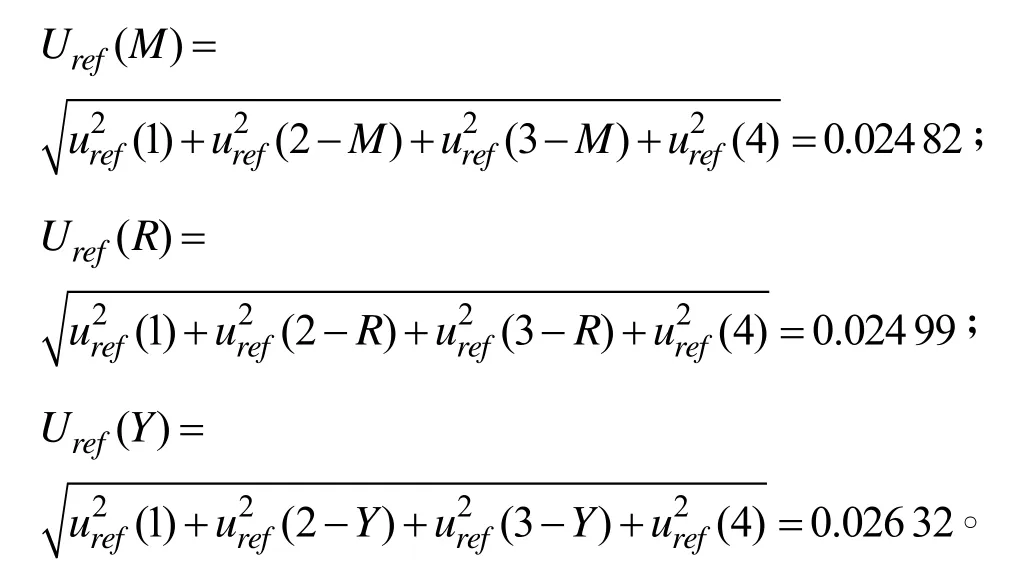
2.5 检测结果的表述
一般情况下,检测结果取包含因子k=2,置信区间95%,表明被测量值的大约95%概率可望包含于此区间,则扩展不确定度U=Uref×k×C0(C0为被测样品含量的平均值,由数学模型获得),因此:
大米试样的扩展不确定度UM=Uref(M)×k×C0(M)=0.024 82×2×8.3=0.5 μg/kg,检测结果表述为大米试样含量XM=(8.3±0.4) μg/kg,k=2;
烟熏肉试样的扩展不确定度UR=Uref(R)×k×C0(R)=0.024 99×2×8.2=0.5 μg/kg,检测结果表述为烟熏肉试样含量XR=(8.2±0.4) μg/kg,k=2;
食用植物油试样的扩展不确定度UM=Uref(M)×k×C0(M)=0.026 32×2×8.7=0.5 μg/kg,检测结果表述为食用植物油试样含量XY=(8.7±0.5) μg/kg,k=2;
3 讨论与结论
通过高效液相色谱法对食品中苯并芘含量测定的不确定度各分量的评定,可见最终检测结果的不确定度主要由标准溶液配制过程、样品前处理过程以及标准曲线拟合引入,其次为高效液相色谱重复测定引入。因此,实际操作过程中,可通过选择纯度较高或者扩展不确定较小的标准溶液、选择精度更高的天平和玻璃量器、增加每个试样的称样次数、优化前处理方法提高样品加标回收率、增加平行样品测定次数等方式来减小不确定度的引入,以提高检测结果的准确性。
此外,本次对大米试样、烟熏肉试样和食用植物油试样进行平行不确定度评定,结果发现电子天平称样过程对食用油的不确定度贡献较其它两种试样大,可能原因是样品称样量最小为0.4 g,其它两种为1.0 g。通过考察三种不同基质样品的加标回收率,发现食用植物油样品的加标回收率最高且引入的不确定度最小,说明食用油基质对回收率的影响最小。从最终检测结果的表述中可以看出三种不同基质的扩展不确定度差别不大。
