气质联用法快速测定肉制品中N-二甲基亚硝胺
2021-01-18侯靖卢跃鹏陈丹王梦颖涂凤琴刘梦婷
侯靖,卢跃鹏,陈丹,王梦颖,涂凤琴,刘梦婷
武汉食品化妆品检验所(武汉 430012)
肉制品是中国人餐桌上广受欢迎的美味,但是其可能存在的亚硝胺污染问题,成为消费者的一块心病。亚硝胺类化合物中受关注最多的是N-二甲基亚硝胺,其为二甲胺和亚硝酸盐作用形成的[1],国际癌症研究所(IARC)将其列为2A级致癌物,即对人类致癌性证据有限,对试验动物致癌性证据充分的致癌物。GB 2762—2017规定肉制品中N-二甲基亚硝胺的限量为3.0 μg/kg。
无论是对肉制品中N-二甲基亚硝胺的污染情况调查,还是对N-二甲基亚硝胺控制方法的研究,均需要快速准确地对肉制品中N-二甲基亚硝胺含量进行检测。常见N-二甲基亚硝胺检测的前处理方法主要有水蒸气蒸馏法[2]、直接溶剂提取法[3]和QuEChERS法[4-5]等。水蒸气蒸馏法取样量较大,操作复杂,且耗时较长,同时前处理过程中的液液萃取易造成待测物的损失,采用同位素稀释的方法可以减少影响,增加定量的准确性[6]。因为同位素标样价格较昂贵,如何减少样品量,从而减少同位素标样的使用,成为需要考虑的问题。直接使用二氯甲烷溶剂提取,方法较为简便,前处理耗时短,且目标物损失少。但是对于含有脂肪的样品,二氯甲烷会同时提取出大量的脂肪,影响检测的准确性,并对仪器造成严重污染。N-二甲基亚硝胺的检测可采用气相色谱-热能分析仪法[7]、气相色谱-质谱法[8]、气相色谱-串联质谱法[2,4-5]和液相色谱-串联质谱法[9]。气相色谱-串联质谱法因检测灵敏度高、准确性好,成为N-二甲基亚硝胺检测的主流方法。
试验报道一种采用超纯水提取、二氯甲烷反萃取的简单前处理方法,结合气相色谱-串联质谱检测,同位素稀释法定量,用于肉制品中N-二甲基亚硝胺的快速检测。方法准确、可靠,可为食品科学研究及食品安全监管提供参考。
1 材料与方法
1.1 仪器与试剂
7890B-7010气相色谱-质谱联用仪(美国Agilent公司);万分之一电子天平(瑞士Mettler Toledo公司);Vortex-Genie 2涡旋混匀器(美国Scientific Industries公司);Direct 8超纯水机(德国Merck公司);超声波清洗机(英国Prima公司)。
N-二甲基亚硝胺(1 000 mg/L,美国O2si公司);N-二甲基亚硝胺-D6(1 000 mg/L,美国O2si公司);二氯甲烷(色谱纯,德国Merck公司)。
1.2 试验方法
1.2.1 标准工作溶液的配制
将N-二甲基亚硝胺与N-二甲基亚硝胺-D6标准溶液用二氯甲烷分别稀释至100和1 000 ng/mL,得到N-二甲基亚硝胺与N-二甲基亚硝胺-D6标准工作溶液。
1.2.2 标准系列溶液的配制
分别吸取5,10,20,50,100,200和500 μL质量浓度100 ng/mL的N-二甲基亚硝胺标准工作溶液,加入5 μL质量浓度1 000 ng/mL的N-二甲基亚硝胺-D6标准工作溶液,定容至1.00 mL,得到质量浓度范围为0.5~50 ng/mL标准系列溶液。
1.2.3 样品处理
称取10 g粉碎后的样品于旋盖离心管中,加入100 μL质量浓度1 000 ng/mL的N-二甲基亚硝胺-D6标准工作溶液,加入20 mL超纯水,旋紧管盖后涡旋,超声提取30 min,冷却至室温后以4 000 r/min离心5 min。吸取2 mL水提取液,注意避免吸到沉淀的样品和水表层的油脂。提取液置于10 mL离心管内,加入2 mL有机溶液,充分涡旋混匀,以4 000 r/min离心5 min,移取有机溶液至进样小瓶中,进行分析。
1.3 仪器分析条件
色谱条件:进样口温度280 ℃;进样模式,不分流进样;进样量2 μL;色谱柱HP-INNOWAX(30 m×0.25 mm×0.25 μm);载气,氦气(纯度>99.999%);载气流量1.0 mL/min;接口温度250 ℃;程序升温,50℃保持1 min,以10 ℃/min升至110 ℃,保持3 min,以40 ℃/min升至230 ℃,保持1 min。质谱条件:离子源温度230 ℃;四极杆温度150 ℃;碰撞能量70 eV;化合物离子对见表1。
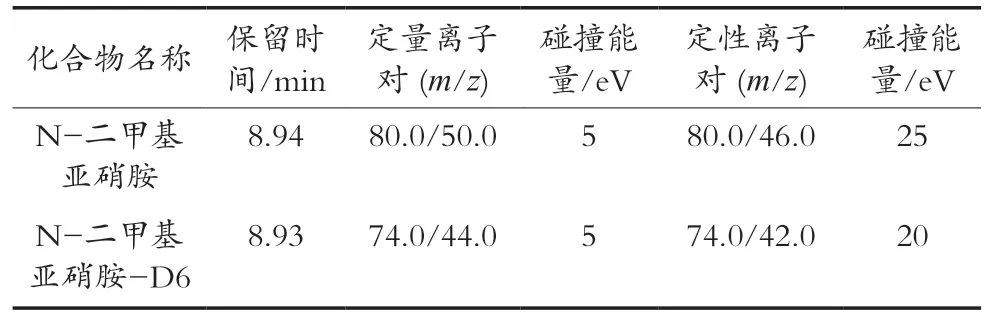
表1 N-二甲基亚硝胺的保留时间和质谱参数
2 结果与讨论
2.1 仪器方法的优化
对于气相色谱,色谱柱的选择与升温程序的优化是获得良好色谱分离的关键。色谱柱选择方法,考察常用的弱极性色谱柱HP-5MS毛细管柱与极性的HP-INNOWAX毛细管色谱柱。试验表明,N-二甲基亚硝胺采用HP-5MS毛细管色谱柱分离时,色谱峰拖尾严重,采用HP-INNOWAX毛细管色谱柱分离时,峰型较好,且峰面积明显大于同等浓度试样在HP-5MS毛细管色谱柱分离时的峰面积,即具有更高的分析灵敏度。因此,选择HP-INNOWAX毛细管色谱柱作为试验用色谱组。对升温程序的优化,使N-二甲基亚硝胺在色谱柱上获得更好的保留,以获得更好的分离效果。质谱参数方面,对N-二甲基亚硝胺及N-二甲基亚硝胺-D6进行全扫描,获得母离子信息,并在不同碰撞能量条件下进行二级质谱扫描,获得子离子信息。选择丰度高、干扰少的子离子,确定N-二甲基亚硝胺及N-二甲基亚硝胺-D6的定量与定性离子对,并对碰撞能量做进一步优化。空白样品与10 μg/kg加标样品N-二甲基亚硝胺定量离子对色谱图见图1。
肉制品基质复杂,蛋白质、脂肪含量高,给前处理带来极大困难。由于N-二甲基亚硝胺极性较大,选择超纯水进行提取,尽可能避免脂肪等非极性物质的干扰。对获得的水提取液,采用等体积的有机溶剂进行反萃取,试验考察乙酸乙酯、二氯甲烷和乙腈反萃取的效果。结果表明,采用乙酸乙酯反萃取时,N-二甲基亚硝胺的绝对回收率较低;使用乙腈反萃取时,N-二甲基亚硝胺的绝对回收率最好,但是干扰物峰也更多,同时使用乙腈反萃取时需要增加盐析的步骤,增加前处理的复杂性。使用二氯甲烷反萃取的回收率也较好,干扰相对较少,故最终选择二氯甲烷进行反萃取。
2.2 线性范围与定量限
将N-二甲基亚硝胺及其同位素内标的标准系列溶液标准溶液进样检测,在0.5~50 ng/mL质量浓度范围内,以N-二甲基亚硝胺与其同位素内标的相对峰面积y和性对浓度x进行线性回归计算。线性方程为y=0.660 5x-0.029 3,相关系数R2=0.999 8,线性良好。在空白肉制品中添加不同量的N-二甲基亚硝胺,按该方法进行处理,以定量离子对性噪比不小于10计算,方法的定量限为1.0 μg/kg。
2.3 回收率与重复性
通过向空白肉制品基质中添加不同量的N-二甲基亚硝胺,分别制备低(1.0 ng/kg)、中(3.0 ng/kg)、高(10 ng/kg)3个浓度水平的空白加标样品,进行回收率试验,结果见表2。结果表明,N-二甲基亚硝胺在3个浓度水平下的平均回收率分别为120.9%,106.6%和97.5%,对应的相对标准偏差分别为5.90%,0.76%和1.51%。方法准确度和精密度良好,符合相关标准要求。

图1 空白样品(a)与10 μg/kg加标样品(b)N-二甲基亚硝胺定量离子对色谱图
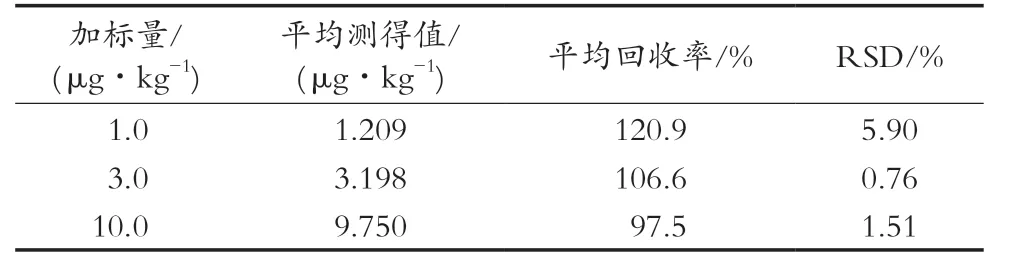
表2 N-二甲基亚硝胺回收率及精密度(n=6)
2.4 实际样品检测
采用该方法对20批次市售肉制品进行N-二甲基亚硝胺含量测定,全部前处理过程耗时不超过2 h。结果显示全部样品均无高出定量限的N-二甲基亚硝胺被检出。
3 结论
建立一种基于同位素稀释-气相色谱-串联质谱测定肉制品中N-二甲基亚硝胺的方法,采用超纯水对样品进行提取,离心分离出水提取液后,用二氯甲烷进行反萃取,有机溶液进样检测。N-二甲基亚硝胺在1.0,3.0和10 μg/kg水平的加标回收率为97.5%~120.9%,相对标准偏差为0.76%~5.90%。相较GB 5009.26—2016《食品安全国家标准 食品中N-亚硝胺类化合物的测定》,方法前处理简单快速,有机溶剂消耗量只有国家标准的1/125,准确度与精密度良好,是一种高效环保的肉制品中N-二甲基亚硝胺检测方法。
