多靶标受体酪氨酸激酶抑制剂的合成与初步活性筛选
2021-01-15单媛媛刘婷婷马瑛王茂义
单媛媛,刘婷婷,马瑛,王茂义
(西安交通大学第一附属医院药学部,陕西 西安 710061)
查尔酮是一种重要的活性天然产物,其结构为1,3-二苯基-2-丙烯基-1-酮,广泛存在于甘草、红花等药用植物[1],具有抗真菌、抗炎、抗肿瘤等生物活性[2],其抗肿瘤活性主要是对受体酪氨酸激酶(receptor tyrosine kinases,RTKs)的抑制[3]。RTK在肿瘤发生、发展中发挥关键的作用,其中,表皮生长因子受体(EGFR)、血管内皮细胞生长因子受体2(VEGFR-2)、成纤维细胞生长因子受体-1(FGFR1)3种RTKs的表达异常与多种肿瘤密切相关,已成为抗肿瘤药物研究的有效靶标[4]。尤为重要的是,这3种RTKs具有高度保守的一级序列和三维结构,其配体结合位点和酪氨酸激酶催化区域高度相似,使得设计同时针对这3种RTKs的多靶标抑制剂成为可能,也有利于解决单靶标抑制剂的耐药性的缺陷[5]。
基于查尔酮为先导化合物,设计并合成新型查尔酮衍生物类多靶标RTKs抑制剂,通过体外活性筛选有望发现具有抗肿瘤活性的新型查尔酮衍生物[6]。基于已报道对RTKs有抑制活性且结构对称的联苯化合物为先导物,首先采用分子剖裂原理将对称的联苯化合物破裂为两分子苯氧基乙酰苯胺结构[7],分析其结构发现:苯氧基乙酰苯胺结构与查尔酮的结构母核类似,而且其结构中不存在α,β-不饱和羰基共轭结构片段,因此不会与体内谷胱甘肽的硫醇发生亲电反应[8],被谷胱甘肽还原,所以保留了查尔酮本身的药效结构特征与生物活性[9]。在此基础上,引入含有氢键供受体网络的苯甲酰胺、溴代苯甲酰胺结构作为RTKs抑制剂的铰链区结合基团[10],并重点考察末端苯胺上不同取代基对活性的影响[11],设计了查尔酮苯甲酰胺系列衍生物(查尔酮-BAA)(见图1)。
1 实验部分
1.1 仪器与试剂 化合物合成所需的试剂按照标准操作流程处理,常用有机溶剂主要购自天津科密欧试剂公司、青岛海洋化工有限公司、国药集团试剂公司等。所用试剂除了有特殊说明之外均为分析纯,合成操作中除水相反应外,其他实验操作按照标准操作程序除水,无氧反应操作均在氮气等惰性气体保护下进行。化合物合成中所有反应通过薄层色谱(TLC)监测反应进程。目标化合物及关键中间体的熔点采用予华仪器有限责任公司的X-4型数显熔点测定仪测定(温度未经校正)。目标化合物的1H-NMR(400 MHz)由西安交通大学理学院核磁共振仪(Bruker Advance AC 400)测定,测试所用氘代溶剂为DMSO-d6和CDCl3,采用四甲基硅烷(TMS)为内标,化学位移值δ单位为ppm。目标化合物的EI-MS由西安交通大学药学院HPLC-MS-QP2010测定。
1.2 方法与结果
1.2.1 化合物的制备[12]基于以上结构优化策略,完成了共35个查尔酮衍生物(查尔酮BAA1~35)的合成(合成路线1与合成路线2)。该系列化合物以异香草醛(1)为起始原料,首先以NaOH/KOH为氧化剂将异香草醛氧化为异香草酸(2),然后将其羟基以苄基保护得到中间体(3),再与氯化亚砜反应生成酰氯,将得到的酰氯分别与甲胺、异丙胺、二乙胺反应得到相应的苯甲酰胺关键中间体(4)。此外,将各种取代苯胺与氯乙酰氯反应得到相应的氯乙酰苯胺(6)。最后将前面制备的苯甲酰胺关键中间体(5)与取代氯乙酰苯胺(6)反应制得相应的查尔酮苯甲酰胺系列衍生物(查尔酮BAA1~21),见图2。
化合物2的合成:称取1.0 g氢氧化钠,1.5 g氢氧化钾加入三颈瓶中,加入0.3 mL蒸馏水,混合物加热至160 ℃使其熔融。分5批缓慢加入3.0 g异香草醛,熔融状态下继续反应10 min,停止加热待反应混合物温度降至室温后,加水100 mL使混合物全部溶解,冰浴条件下用2 mol·L-1盐酸将混合物调pH至1~2。混合物溶液直接抽滤,用水洗涤滤饼2~3次既得产物,将产物放入真空干燥箱干燥。
化合物3的合成:化合物2(3.47 g,15 mmol)溶于70 mL无水乙醇中,加入无水碳酸钾(6.22 g,45 mmol)和氯化苄(2.30 mL,20 mmol),回流反应6 h,除去碳酸钾,减压蒸除乙醇,乙酸乙酯(30 mL×3)提取,分别用H2O (3×30 mL),2 mol·L-1NaOH 水溶液(3×30 mL),2 mol·L-1HCl 水溶液(3×30 mL),饱和氯化钠水溶液 (2×30 mL)洗涤,无水硫酸钠干燥,柱层析分离得到产物(4.56 g,95%) 。
化合物4的合成:将化合物3(6.72 g,26 mmol)溶于10 mL无水二氯甲烷中,冰浴条件下缓慢滴加5.82 mL氯化亚砜的二氯甲烷溶液和5滴催化量的二甲基甲酰胺(DMF),室温条件下3 h。待反应完全后减压蒸除溶剂,将产物溶于无水二氯甲烷中备用。冰浴条件下,将上述酰氯的无水二氯甲烷溶液(25 mL)缓慢逐滴加入22.91 mL 30%的甲胺水溶液中,冰浴反应2 h后升温至室温反应。反应完全后,加入10 mL水,分液,水相用二氯甲烷(3×30 mL)提取,合并有机相,有机相分别用2 mol·L-1的盐酸(3×25 mL),水(2×25 mL),饱和碳酸氢钠(2×25 mL),饱和氯化钠(2×20 mL)洗涤,无水硫酸钠干燥,柱层析分析得到产物(3.16 g,84%)。
化合物5的合成:化合物4(3.49 g,10 mmol)溶于100 mL无水甲醇中,加入10%的Pd/C(0.10 g)。通入氢气还原反应,室温条件下搅拌反应。反应完全后,过滤,回收Pd/C,减压蒸除甲醇,得到产物(2.58 g,97%)。
目标化合物(BAA1)的合成:将化合物5(1.58 g,5.90 mmol)溶于70 mL无水乙醇中,加入无水碳酸钾(2.45 g,17.70 mmol)。反应混合物室温条件下搅拌反应30 min,加入化合物6 (1.54 g,6.49 mmol)。混合物回流反应10 h,TLC监测反应进程,待反应完全后,过滤除去碳酸钾,减压蒸除乙醇,乙酸乙酯(3×20 mL)提取,有机相依次用H2O (3×10 mL)、2 mol·L-1NaOH(3×10 mL)、2 mol·L-1HCl (3 ×10 mL) 和饱和氯化钠水溶液 (2×10 mL)洗涤,无水硫酸钠干燥。柱层析分离得到目标化合物BAA1(2.13 g,78%)。mp:197~198 ℃。EI-MS(m/z):366.1 (M+)。1H-NMR (400 MHz,CDCl3) δ 8.88 (s,1H),7.80 (d,J=4.7 Hz,1H),7.51(s,1H),7.46(d,J=8.4 Hz,2H),7.17-7.12 (m,1H),6.97 (d,J=8.4 Hz,1H),6.27 (s,1H),4.69 (s,2H),4.00(s,3H)。3.03 (s,3H)。化合物BAA2~21的制备方法与BAA1相同。
查尔酮衍生物(查尔酮BAA22~35)的合成见合成路线2,该系列化合物同样以异香草醛为起始原料,首先以液溴为溴化试剂制备溴代异香草醛,将其羟基以苄基保护,然后以次氯酸钠为氧化剂将醛基氧化为羧基制备溴代异香草酸。再与氯化亚砜反应生成酰氯,将得到的酰氯分别与甲胺、异丙胺、二乙胺反应得到相应的苯甲酰胺关键中间体。此外,将各种取代苯胺与氯乙酰氯反应得到相应的氯乙酰苯胺。最后将前面制备的苯甲酰胺关键中间体与取代氯乙酰苯胺反应制得相应的查尔酮苯甲酰胺系列衍生物(查尔酮BAA22~35)(见图3)。
1.2.2 体外抗肿瘤活性筛选[13-14]体外筛选了目标化合物对EGFR(表皮生长因子受体)、VEGFR-2(血管内皮细胞生长因子受体-2)和FGFR1(成纤维细胞生长因子受体-1)3种受体酪氨酸激酶的抑制活性;细胞水平筛选了目标化合物对乳腺癌细胞(MCF-7)、非小细胞肺癌细胞(A549)、白血病细胞(K562)3种肿瘤细胞的增殖抑制活性[14];寻找基于EGFR/VEGFR-2/FGFR1多靶标的抗肿瘤候选化合物。目标化合物对受体酪氨酸激酶抑制活性筛选所用的试剂盒为ADP-Glo Kinase Assay检测试剂盒(Promega),实验对照药物选用吉非替尼(Gefitinib);肿瘤细胞增殖抑制活性筛选采用经典的噻唑蓝(MTT)方法,对照药物选用吉非替尼(Gefitinib)。
查尔酮衍生物对EGFR、VEGFR-2和FGFR1 3种RTI抑制活性结果(见表1)可以看出,与阳性药吉非替尼相比,大部分化合物对EGFR、VEGFR-2和FGFR1具有一定程度的抑制活性。对EGFR抑制活性中,化合物BAA22、BAA23、BAA30和BAA31的活性结果接近(IC50小于20 nmol·L-1)吉非替尼(IC50=4.07 nmol·L-1),这4个化合物的结构特征是结构母核为溴代苯甲酰胺结构,末端苯胺为3-氯-4-氟苯胺或者3-三氟甲基苯胺;化合物BAA25对EGFR无抑制活性,说明溴代苯甲酰胺是EGFR的有效结构片段,而3-氯-4-氟苯胺或者3-三氟甲基苯胺对EGFR的抑制活性是必需的;有11个化合物的IC50值在1~30 nmol·L-1,其中有4个化合物的活性在20 nmol·L-1以下,值得深入研究。对于VEGFR-2,4个化合物的活性结果较好,分别是BAA10(IC50=17.09 nmol·L-1),BAA16(IC50=14.65 nmol·L-1),BAA19(IC50=15.50 nmol·L-1)和BAA30(IC50=12.44 nmol·L-1);对于FGFR1,目标化合物的抑制活性普遍低于对EGFR和VEGFR-2的抑制活性,说明该系列化合物对FGFR1的选择性不如EGFR和VEGFR-2,其次,化合物BAA1(IC50=19.54 nmol·L-1),BAA3(IC50=13.69 nmol·L-1)和BAA29(IC50=19.23 nmol·L-1)的活性较好。从整体活性结果显示,化合物BAA30对EGFR、VEGFR-2和FGFR1的抑制活性均较好,特别是对EGFR和VEGFR-2的抑制活性明显高于其他化合物,说明BAA30结构中的溴代苯甲酰胺是较为理想的多靶标酪氨酸激酶抑制剂铰链区结合基团,其末端的3-三氟甲基取代基对于保持化合物的多靶标抑制活性是必需的。
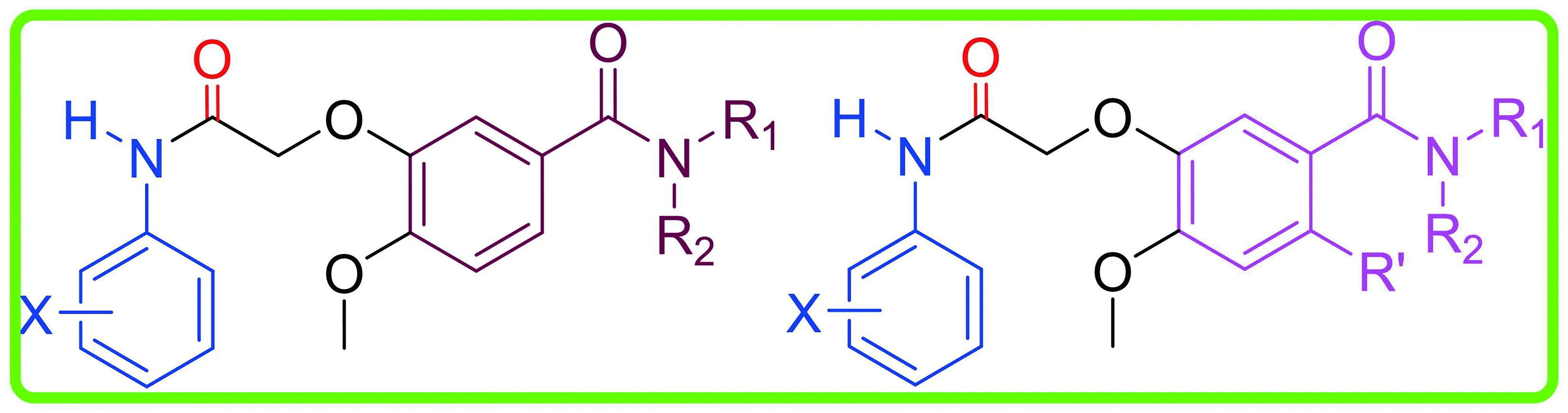
表1 查尔酮衍生物对受体酪氨酸激酶的抑制活性结果(IC50,nmol·L-1)

表1(续)
查尔酮衍生物对乳腺癌细胞(MCF-7)、非小细胞肺癌细胞(A549)、白血病细胞(K562)的增殖抑制活性见表2。大部分衍生物对3种肿瘤细胞均具有一定的增殖抑制活性,其中活性最高的化合物是BAA23,其结构特征为苯甲酰胺母核上2-位为溴取代基,末端苯胺为3-三氟甲基苯胺,对MCF-7、A549、K562的抑制活性IC50值分别为5.42、8.27、9.82 μmol·L-1。对于不含溴的苯甲酰甲胺类衍生物,活性最高的是BAA5(末端为4-溴苯胺),对3种肿瘤细胞增殖抑制活性IC50值分别为12.7、15.2、13.3 μmol·L-1。对于不含溴的苯甲酰异丙胺类衍生物,活性最高的是BAA8(末端为3-三氟甲基苯胺),对3种肿瘤细胞增殖抑制活性IC50值分别为21.7、10.4、8.93 μmol·L-1;该系列中对MCF-7抑制活性最高的是BAA14(末端为3-氟苯胺)。对于不含溴的苯甲酰二乙胺类衍生物,活性最高的是BAA16(末端为3-三氟甲基苯胺),对3种肿瘤细胞增殖抑制活性IC50值分别为24.1、20.4、12.4 μmol·L-1。对于苯甲酰胺母核上含溴取代基的化合物(BAA22~BAA35),其抗肿瘤活性普遍优于不含溴的衍生物,包括活性最高的化合物BAA23。苯甲酰甲胺类衍生物中BAA26和BAA27对3株肿瘤细胞的增殖抑制活性值在20~30 μmol·L-1之间,活性较好。苯甲酰异丙胺系列化合物中,BAA33和BAA36的活性较好,与BAA26接近。总结苯甲酰胺系列衍生物构效关系发现:苯甲酰胺母核上溴取代基有利于提高活性,末端苯胺上的三氟甲基与氟取代基有利于抗肿瘤活性。
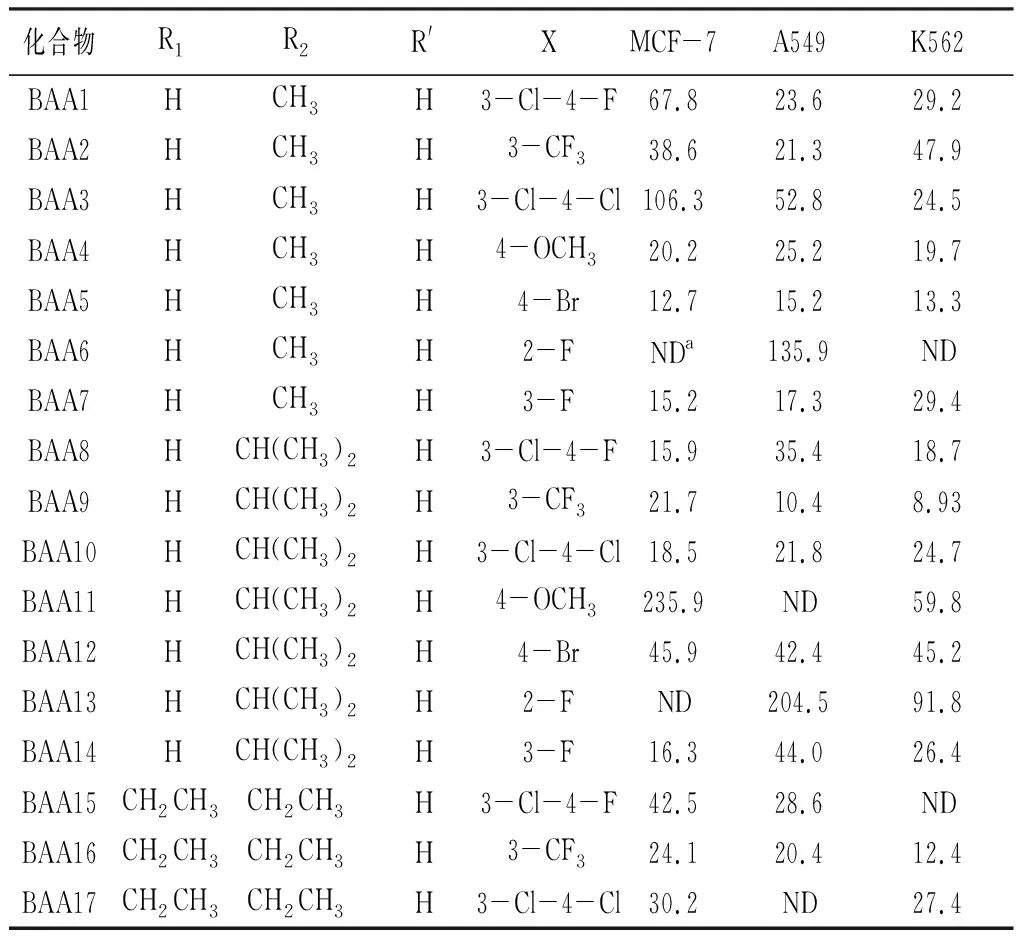
表2 查尔酮衍生物对不同肿瘤细胞株的抗增殖活性(IC50,μmol·L-1)

表2(续)
2 讨论
以查尔酮为先导化合物,针对查尔酮母核结构中α,β-不饱和双键的缺陷,共设计、合成了35个新型查尔酮衍生物。合成路线合理,反应条件比较温和,均由廉价易得的异香草醛为原料制备。体外活性实验表明,大部分化合物具有较好的RTK抑制与抗肿瘤活性,部分化合物的活性接近阳性对照的水平。为新型查尔酮先导化合物的结构优化提供了依据。
