马铃薯S病毒RT-qPCR通用检测体系的建立与应用
2021-01-08程胜群吕文河高艳玲白艳菊范国权邱彩玲董学志白雅梅
程胜群,吕文河,高艳玲,,白艳菊,范国权,张 威,张 抒,邱彩玲,申 宇,董学志,白雅梅
(1.东北农业大学 农学院,黑龙江 哈尔滨 150030;2.黑龙江省农业科学院 马铃薯研究所,黑龙江 哈尔滨 150086;3.东北农业大学 资源与环境学院,黑龙江 哈尔滨 150030)
作为中国第四大粮食作物,马铃薯产量高、营养丰富,在确保粮食安全方面具有重要作用[1]。预计到2020年马铃薯种植面积可以扩大到667万 hm2[2]。但由于病毒病的危害,导致产量和品质下降。侵染马铃薯的病毒大约有40种[3],马铃薯S病毒(PotatovirusS, PVS)是影响全球马铃薯生产仅次于马铃薯Y病毒(PotatovirusY, PVY)和马铃薯卷叶病毒(Potatoleafrollvirus, PLRV)的第三大病毒[4]。2004-2009年,PVS在中国马铃薯主产区大田种薯的检出率为0.32%~19.80%,仅次于PVY[5-8],带毒率由北向南呈增高趋势[9]。
PVS属于乙型线形病毒科(Betaflexviridae)香石竹潜隐病毒属(Carlavirus)[10],是长610~710 nm的弯曲线状粒子,正单链RNA(+ssRNA)长8.4~8.5 kb[11]。PVS于1948年在荷兰首次发现[12],遗传进化分析表明,PVS起源于南美,后被引入欧洲并扩展为主要传播中心[13],目前广泛分布于美国、英国、荷兰、伊朗等马铃薯产区[14]。根据PVS侵染昆诺瓦藜(Chenopodiumquinoa)的症状可将其分为PVSO(Ordinary strain)和PVSA(Andean strain)株系[9],其中,PVSO侵染后昆诺瓦藜表现局部枯斑,而PVSA株系则表现为系统侵染。Cox等[15]发现并建议将昆诺瓦藜表现系统侵染但与PVSO遗传关系较近的分离株命名为PVSO-CS(CS=Chenopodiumsystemic)。并建议将来使用PVSA-CL(CL=Chenopodiumlocalized)代表不能系统侵染昆诺瓦藜但与PVSA遗传关系较近的分离株。2018年,欧洲发现5个PVS重组分离株,其中1个为PVSA-CL[16]。此外,PVS分离株RVC在全序列遗传进化树中既不属于PVSO,又不属于PVSA,可能代表了一种新的PVS株系[11]。目前,PVSO和PVSA在中国内蒙古、黑龙江、辽宁、河北、山东、青海、广西、湖南、四川、浙江、福建、贵州等地均有报道[17]。
PVS可通过机械摩擦和蚜虫传播,种植在田间的马铃薯原种能迅速感染PVS,一个生产季后感染率可达70%[18]。黄萍等[19]对大西洋原原种连续繁种的生产试验进行4种病毒检测,PVS的检出率最高,种植1 a后带毒率为3%,种植3 a后带毒率高达30%。PVS在大多数马铃薯品种中症状不明显[9],只在易感品种中表现叶脉深陷、叶片皱缩、小的坏死斑点等症状[20]。与PVSO相比,PVSA能引起更严重的症状[21],在指示植物昆诺瓦藜中引起系统坏死斑[9]。PVS与其他病毒复合侵染时可加剧马铃薯症状[22],如PVS与马铃薯X病毒(PotatovirusX, PVX)复合侵染可增加植株中PVS的浓度,导致马铃薯出现重花叶症状[23]。PVS单独侵染导致马铃薯减产10%~20%[24],而与其他病毒复合侵染时可增至30%[22]。由于PVS单独侵染在马铃薯上引起的症状较轻,因此在种薯生产中难以防控。2004-2009年,在试管苗中检出率最高,为18.94%~36.69%[5-8,19],这可能与PVS在茎尖脱毒中比较难以脱除有关[25-26]。试管苗是脱毒种薯生产的源头,因此,在种薯生产中对茎尖剥离材料、试管苗、原种及各级种薯进行PVS检测具有重要意义。
目前,常规检测病毒的方法有双抗体夹心酶联免疫吸附法(Double antibody sandwich enzyme-linked immunosorbent assay, DAS-ELISA)、反转录聚合酶链式反应(Reverse transcription polymerase chain reaction, RT-PCR)、实时荧光定量PCR(Real-time fluorescent quantitative PCR, RT-qPCR)等。DAS-ELISA法适合检测叶片组织,但存在耗时长、灵敏度低等问题,而RT-qPCR方法灵敏度高,是常规RT-PCR法的10倍以上[27]。此外,RT-qPCR方法还具有操作简单,特异性强,准确度高等优点。目前,已经建立了PVX[28]、PVY[29]、PVS[30]、马铃薯A病毒(PotatovirusA, PVA)和PLRV[31]的RT-qPCR检测体系。
RT-qPCR法自出现以来,已广泛应用于病毒、真菌、转基因等检测及遗传育种领域[32]。但随着GenBank中PVS序列逐渐增多,PVS显示出丰富的遗传多样性,现有PVS检测引物不能与之高度匹配,未能检出一些特殊株系和未分株系。本试验比对分析了NCBI GenBank中现有PVS序列,设计PVSO和PVSA的通用引物,并可根据扩增片段熔解温度(Melting temperature, Tm)鉴定2种株系。此外,通过构建的标准曲线并测试检测体系灵敏度,测定感染PVS的马铃薯叶片、叶柄、茎、根和休眠块茎中病毒含量,确定适合检测的马铃薯组织类型,尤其是休眠块茎。本研究旨在建立一套快速、准确的PVS RT-qPCR检测体系,并可方便快捷的鉴定PVSO和PVSA株系。可直接检测马铃薯休眠块茎等不同部位的样品,为茎尖脱毒材料筛选、试管苗病毒筛查、脱毒种薯生产等提供技术支持。
1 材料和方法
1.1 试验材料
供试毒源及植物:感染PVSO的马铃薯品种尤金试管苗G89和感染PVSA的试管苗G93,以及分别感染 PVX、PVY、马铃薯M病毒(PotatovirusM, PVM)、PVA 和 PLRV 的马铃薯试管苗G84、G87、G98、G35和G76,均由黑龙江省农业科学院马铃薯研究所保存。健康马铃薯试管苗G1亦由该所提供。
1.2 试验方法
1.2.1 植物总RNA的提取及cDNA的合成 RNA的提取:分别切取马铃薯试管苗G89、G93、G84、G87、G98、G35和G76和健康试管苗G1的茎叶100 mg,放于-20 ℃预冷的研钵中,加入液氮充分研磨后迅速转移至1.5 mL 离心管中,加入1 mL TRIzol,按照TRIzol试剂说明书方法提取后的植物总RNA放于-80 ℃冰箱保存。
cDNA合成:取4 μL RNA、1 μL随机引物、5 μL DNA/RNA酶去离子水,70 ℃预变性5 min,冰上放置2 min。依次加入5×反转录缓冲液5 μL、10 mmol/L dNTP 1.25 μL、40 U/μL RNA酶抑制剂 0.5 μL、200 U/μL M-MLV反转录酶1 μL、无DNA/RNA酶去离子水7.25 μL。反转录程序为37 ℃ 1 h,92 ℃ 5 min。合成的cDNA保存于-20 ℃冰箱备用。
1.2.2 RT-qPCR检测体系的建立 引物设计:下载NCBI数据库GenBank中所有PVS序列,应用软件BIo-edit进行比对分析后,以来自中国的PVSO分离株HB24(GenBank登录号为 KU896945)和PVSA分离株HB7(GenBank登录号为KU896946)为模板,应用Primer 5.0在保守区域CP(Coat protein,CP)基因处设计引物,PVS-F序列为5′-AAAGTGTG CAGGCTGTATGC-3′,在两序列上位置为7 761~7 780 bp,PVS-R序列为5′-ATCTCAGCGCCRAGC AT-3′,在两序列上位置为8 046~8 062 bp,扩增目的片段长度为302 bp,PVSO株系目的片段Tm理论值为88.9 ℃,PVSA株系Tm理论值为89.7 ℃,相差0.8 ℃。引物合成及序列测定委托生工生物工程(上海)股份有限公司完成。
RT-qPCR 20 μL反应体系:10×PCR缓冲液2 μL、25 mmol/L MgCl22 μL、2.5 mmol/L dNTP 0.8 μL、10 μmol/L上下游引物各0.4 μL、Eva-Green 1 μL、5 U/μLTaq酶 0.12 μL、cDNA 2 μL,用无DNA/RNA酶去离子水补至20 μL。反应程序:95 ℃ 15 min;95 ℃ 20 s,56 ℃ 20 s,72 ℃ 30 s,采集荧光信号,40个循环;60~95 ℃分析熔解曲线。实时荧光定量PCR仪型号为Roche LightCycler® 480。
1.2.3 RT-qPCR特异性检测 马铃薯试管苗G89、G93、G84、G87、G98、G35和G76的cDNA为模板,以无DNA/RNA酶去离子水为空白对照,健康试管苗G1的cDNA为阴性对照进行RT-qPCR扩增,根据熔解曲线判断有无非特异扩增。
1.2.4 质粒标准品制备、标准曲线建立和灵敏度检测 以马铃薯试管苗G89和G93的cDNA为模板,以PVS-F和PVS-R为引物进行RT-PCR扩增,反应程序为95 ℃ 3 min;95 ℃ 30 s,56 ℃ 30 s,72 ℃ 45 s,72 ℃ 10 min, 4 ℃ 5 min。经1.5% 琼脂糖电泳检测后回收PCR产物,与pESI-T载体连接,转化到DH5α感受态细胞中,用RT-PCR法鉴定阳性克隆,应用质粒小提试剂盒提取重组质粒并测序。质粒拷贝数=6.02×1023×质粒浓度(g/μL)/(载体长度+片段长度)×660,式中6.02×1023为阿佛伽德罗常数,载体长度为1 865 bp,片段长度为302 bp,用紫外分光光度计测试重组质粒浓度,计算得到拷贝数,将重组质粒按照拷贝数进行10倍梯度稀释成标准品进行RT-qPCR扩增,每个模板3次重复,以起始模板数的对数为X轴,荧光信号基线的循环数平均值(Cycle threshold,Ct值)为Y轴作回归曲线,构建标准曲线,将标准品倍比稀释成8个浓度梯度检测灵敏度,同时进行常规PCR扩增,测定检测灵敏度。
1.2.5 田间马铃薯样品检测 马铃薯样品随机采自黑龙江、吉林、辽宁、河北、陕西、四川、云南、贵州、广东、重庆、内蒙古11个省(市、自治区)的马铃薯生产田,应用PVS DAS-ELISA检测试剂盒检测,筛选90份PVS阳性样品,用所建的RT-qPCR检测体系进行验证。
1.2.6 马铃薯植株5个部位PVS含量测定 马铃薯品种尤金和冀张薯12号原种种植于温室盆钵中,株高20 cm时,分别接种PVSO和PVSA毒源样品G89和G93。接种30 d后,采集马铃薯叶片、叶柄、茎和根,分别在上述部位取100 mg组织,液氮处理后提取植物总RNA。收获后,采集马铃薯休眠块茎提取总RNA,5个部位RNA用水稀释到100 ng/μL。应用PVS RT-qPCR检测体系进行检测,根据所建的标准曲线测定各部位PVS含量。对2个品种尤金和冀张薯12号分别进行统计分析,按二因素(PVS株系和不同部位)完全随机设计实施,3次重复,采用新复极差法进行处理平均值的多重比较。
2 结果与分析
2.1 RT-qPCR检测体系的建立
PVSO分离株G89和PVSA分离株G93扩增后,PCR产物熔解曲线温度为60~95 ℃,分别在85.77~86.00 ℃和87.78~87.91 ℃出现特异熔解峰,Tm值不同,平均数相差1.99 ℃(图1)。

左侧85.77~86.00 ℃为样品G89的熔解峰;右侧87.78~87.91 ℃为样品G93的熔解峰。
2.2 特异性检测
PVSO马铃薯试管苗G89扩增后熔解曲线在86.72 ℃出现特异峰,PVSA马铃薯试管苗G93扩增后熔解曲线在88.05 ℃出现特异峰;而感染PVX、PVY、PVM、PVA和PLRV的阳性样品,以及阴性对照和空白对照扩增后未出现此特异熔解峰(图2),说明该体系特异性好。在78.03 ℃出现的较低熔解峰为引物二聚体的熔解峰,与特异熔解峰相距较远,不影响结果判读。
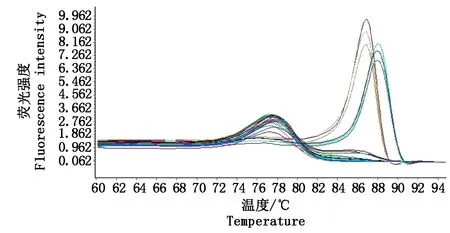
左侧86.72 ℃为G89的熔解峰;右侧88.05 ℃为G93的熔解峰;在86~89 ℃无熔解峰的样品为G84、G87、G98、G35、G76、G1和水。
2.3 标准曲线建立及灵敏度检测
PVSO、PVSA重组质粒浓度分别为26.00,29.86 ng/μL,经计算PVSO拷贝数为1.09×1010拷贝/μL,PVSA拷贝数为1.26×1010拷贝/μL。PVSO、PVSA起始模板量的对数值与Ct值的线性关系见图3,4,PVSO质粒浓度在1.09×105~1.09×109拷贝/μL间呈明显的线性关系,Y=-3.459lgX+37.94(决定系数为R2=0.994 2),熔解曲线在85.93 ℃出现特异峰;PVSA质粒浓度在1.26×105~1.26×109拷贝/μL间呈明显的线性关系,Y=-3.522lgX+40.20(决定系数为R2=0.991 2),熔解曲线在87.91 ℃出现特异峰,其中X为模板拷贝数,Y为样品循环数,Ct值与DNA拷贝数对数之间的线性关系良好,2个重组质粒作为荧光定量PCR检测的标准品是合格的。分别以PVSO、PVSA稀释后的质粒标准品为模板检测灵敏度,结果表明,灵敏度分别为1.09×103,1.26×103拷贝/μL(图5,6),常规PCR可检测1.09×105,1.26×105拷贝/μL,RT-qPCR灵敏度是常规PCR的100倍。
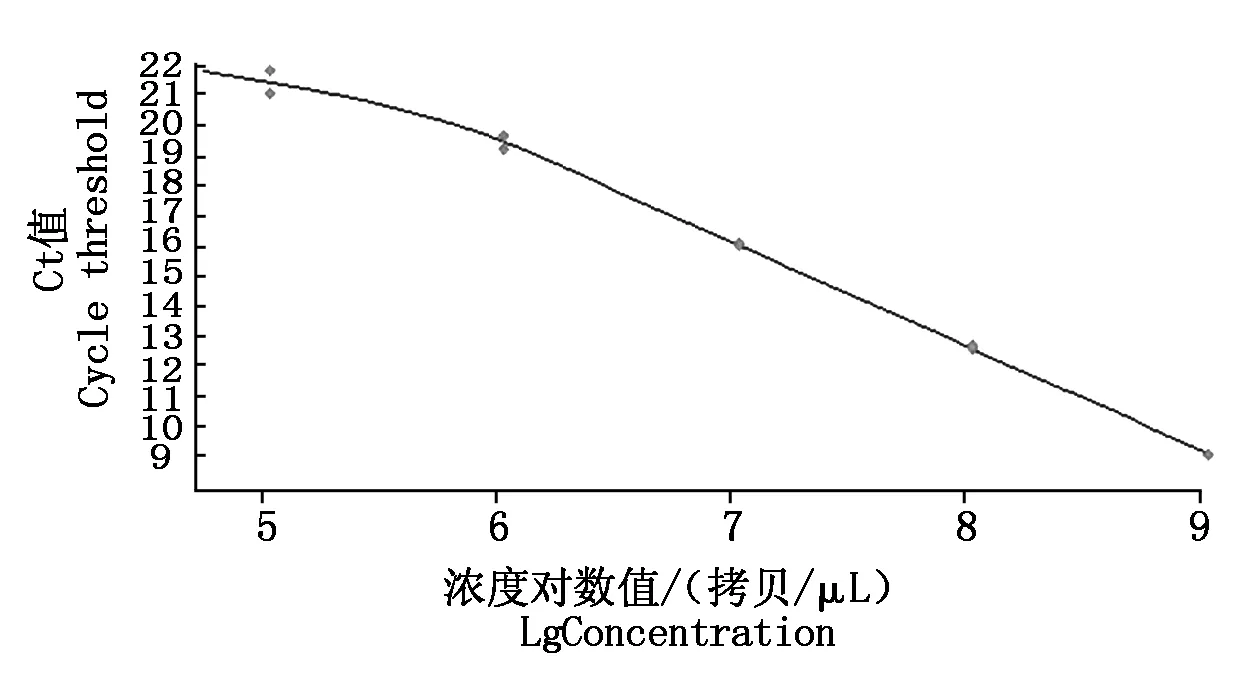
图3 PVSO RT-qPCR标准曲线

图4 PVSA RT-qPCR标准曲线
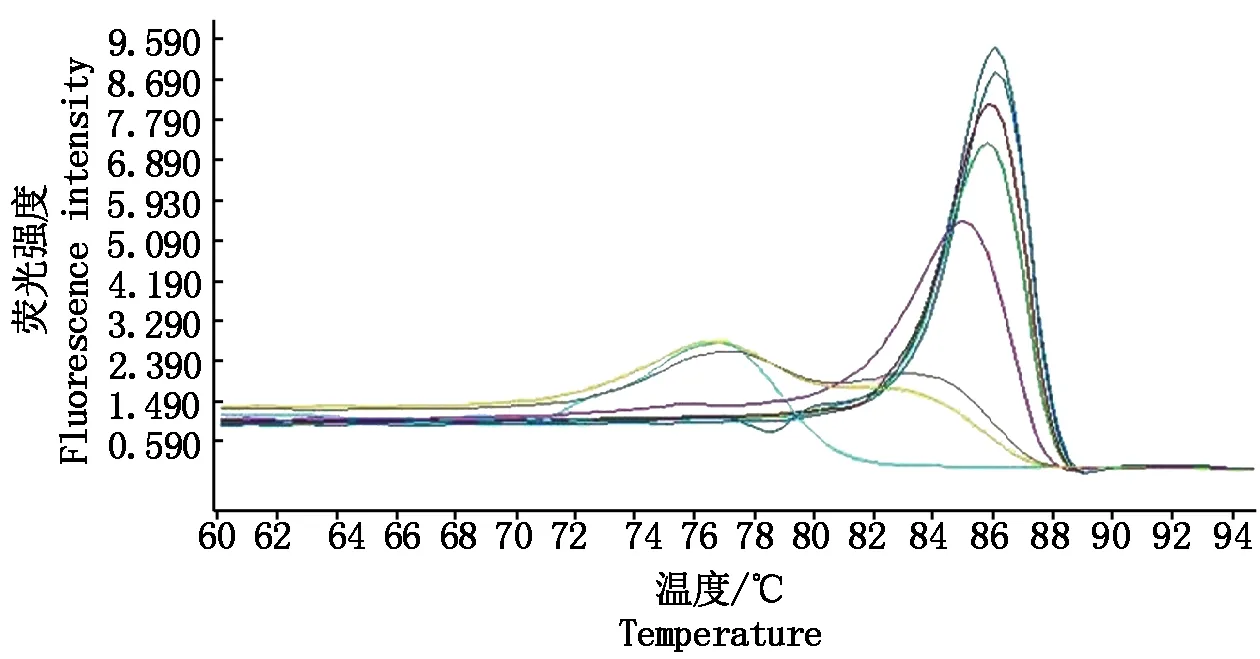
荧光值由高到低依次为重组质粒1.09×108,1.09×107,1.09×106,1.09×105,1.09×104,1.09×103拷贝/μL和水的熔解曲线。
2.4 田间样品检测结果
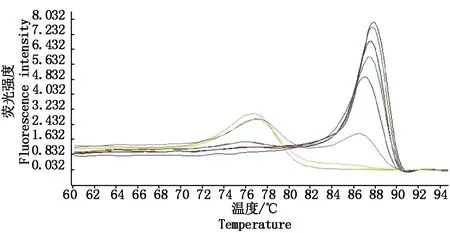
来源于11个省(市、自治区)马铃薯田的90个样品扩增后,熔解曲线在85.82~87.95 ℃出现3组特异峰(图7),与DAS-ELISA检测结果和测序结果相符。不同样品扩增产物的Tm值相差0.86~2.13 ℃。其中Tm在85.82~86.10 ℃是PVSO株系,86.96~87.95 ℃是PVSA株系,在86.23~86.63 ℃出现特异峰的为同时感染PVSO和PVSA的样品。马铃薯中PVSO含量在9.08×105~5.92×107拷贝/μL,马铃薯中PVSA含量在1.67×107~2.15×108拷贝/μL。
荧光值由高到低依次为重组质粒1.26×108,1.26×107,1.26×106,1.26×105,1.26×104,1.26×103拷贝/μL和水的熔解曲线。

图7 大田马铃薯样品熔解曲线
2.5 马铃薯植株5个部位PVS含量测定
5个部位组织中PVS病毒含量(表1)均在107及以上数量级。PVSO在尤金中5个部位每微升RNA中病毒拷贝数从高到低依次是叶柄、叶、根、茎和块茎,而冀张薯12号根比叶含量高,但二者差异不显著。PVSA在尤金的叶柄、叶、根和薯块中病毒拷贝数在2.71×107(茎)~4.00×107(叶柄),但部位间差异均不显著;PVSA在冀张薯12号根中含量最高,为7.45×107拷贝/μL,与叶柄差异不显著,二者显著高于其在叶片、块茎和茎中的含量。
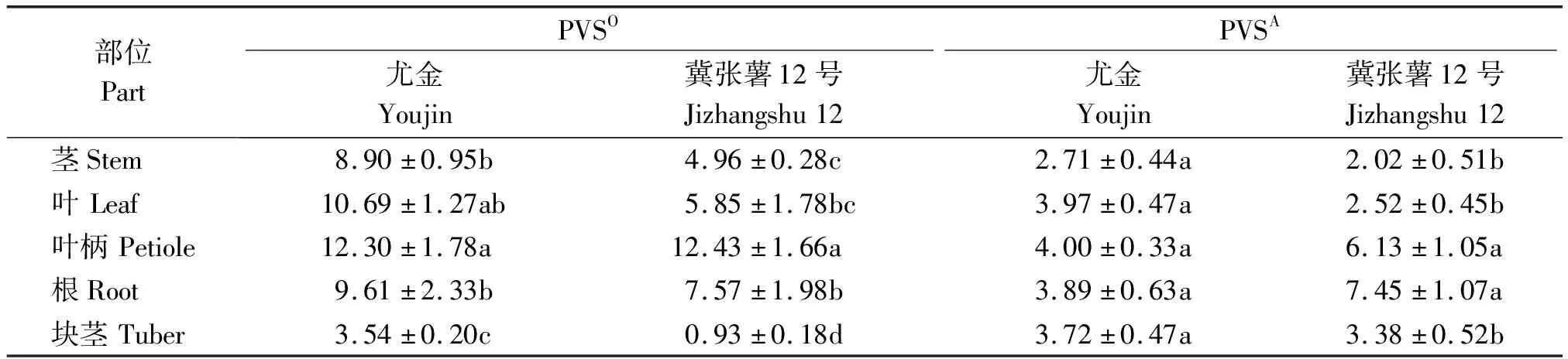
表1 尤金和冀张薯12号5个部位PVSO和PVSA含量
3 讨论与结论
合格的种薯是保证马铃薯高产、优质、高效生产的前提,因此,对马铃薯病毒进行高效、灵敏、快捷检测就显得尤为重要。生物学方法周期长,不适用于大量样品,且指示植物的症状易受环境影响,还需结合血清法进行判断。血清学DAS-ELISA法适合样品量较大的田间样品,不适用于病毒含量较低的休眠薯块以及茎尖脱毒后生长的微量试管苗材料,此外,亦不能区分PVS株系。RT-qPCR技术弥补了这些不足,是一种高效、灵敏、快捷的检测方法,并已用于多种病毒的检测。
PVS序列变异较大,PVSO分离株HB24和PVSA分离株HB7的序列同源性为81.92%(经NCBI Blast 比对)。本试验在两序列保守区域CP基因处设计引物,经比对上游引物(PVS-F)仅在两序列7 763位碱基不同,在引物5′端第3位,未作处理。下游引物(PVS-R)仅在两序列8 051位碱基不同,应用了1个简并碱基R(R=A/G)来提高检测准确性。经比对分析,引物与GenBank中登录的PVSA和PVSO株系有较好的匹配度。其中,PVSO-CS序列遗传进化分析结果表明,3个PVSO-CS株系中,2个属于PVSO第1亚组,1个属于PVSO第7亚组[15],并未独立于PVSO之外,且PVSA-CL序列位于PVSA组中[16]。此外,本试验设计的PVS通用引物已避开重组分离株的重组位点6 088,2 633,5 858,7 204 nt,匹配度较好。同时,PVSA和PVSO株系目的片段碱基差异较大,如HB24和HB7的目的片段理论Tm值相差0.8 ℃,因此,通过分析熔解曲线,不仅检测到PVS病毒,而且可区分2个株系,中国90个PVS分离株Tm值差异为0.86~2.13 ℃。此外,该体系检测特异性强,对于马铃薯上常发生的PVX、PVS、PVA、PVM和PLRV阳性样品无扩增,保证了检测的准确性。
不同地域之间的PVS序列亲缘关系较远[13],因此需要考虑不同地域的PVS分离株扩增情况。目前,中国PVS序列报道仅有29条,采自于河北、广东、广西、浙江和云南,未涵盖我国主要的马铃薯种薯产区。因此,应用来源于11个省(市、自治区)的90个经DAS-ELISA检测感染PVS的叶片样品,用于验证该检测体系的实用性,检测结果与DAS-ELISA法及测序结果一致,进一步说明所建体系具有准确性。而且马铃薯叶片中PVSO含量在9.08×105~5.92×107拷贝/μL,PVSA含量在1.67×107~2.15×108拷贝/μL,均在检出限以上,因此该检测体系具有较强的实用性。目前,多使用DAS-ELISA法检测大田样品病毒,但易出现假阴性。宋静静等[33]用ELISA方法在109个样品中只检测出4个PVS阳性样品,而用RT-PCR则能检出29个。在本研究中,可能是由于在夏季传播PVS的蚜虫更适宜生长,因而夏季采集的样品PVS含量较高,因此DAS-ELISA法未出现假阴性,与RT-qPCR法检测结果一致。此外,本研究中叶片样品PVS含量较高也可能是DAS-ELISA与RT-qPCR检测结果一致的原因。由于使用了简并引物,该体系检测灵敏度虽然比Cheng等[30]报道的低10倍,但准确性高。PVS变异较大且表现隐症,出现漏检将对马铃薯种薯生产产生重大危害。另外,能同时检测出2种株系也为了解我国PVS株系发生特点及遗传多样性研究打下基础。该体系能准确检测马铃薯5种组织中的PVS。PVX 和PVS 是马铃薯茎尖脱毒工作中最难脱除的病毒,因此,最好的办法是应用该体系直接检测备选休眠块茎,剔除感染PVS的块茎,由于该方法灵敏度足以检测休眠块茎,因此,不必把块茎进行催芽种植。而且该体系可检测100 mg组织,对于检测茎尖脱毒后的生长的微小试管苗样品尤为适用,总之,可加速脱毒试管苗的剥离和筛选过程。PVSO在马铃薯叶片、叶柄、茎、根中的含量高于PVSA,但是休眠块茎中相差不大;2个马铃薯品种5个部位PVS病毒拷贝数均大于107数量级,是RT-qPCR检出限的10 000倍,因此,叶片、叶柄、茎、根和休眠块茎等5个部位组织均可用于检测,可根据实际情况和需求,选择相应的部位进行检测。
本研究针对PVS CP基因设计了一对通用简并引物,建立了快速、准确、特异的RT-qPCR检测体系,可依据样品熔解曲线鉴定PVSO和PVSA株系,该方法在马铃薯品种叶片、叶柄、茎、根和休眠块茎等5个部位中检出的PVS病毒拷贝数均大于107数量级,是RT-qPCR检出限的10 000倍,准确率高,实用性强,可为生产脱毒种薯提供技术支持。
