甘草“调和诸药”生物药剂学机制的研究进展
2021-01-06罗子宸单进军狄留庆
罗子宸,张 雯,杨 瑞,单进军*,狄留庆*
甘草“调和诸药”生物药剂学机制的研究进展
罗子宸1, 2, 3,张 雯1, 2,杨 瑞3,单进军3*,狄留庆1, 2*
1. 南京中医药大学药学院,江苏 南京 210023 2. 南京中医药大学 江苏省中药高效给药系统工程技术研究中心,江苏 南京 210023 3. 南京中医药大学 江苏省儿童呼吸疾病(中医药)重点实验室,江苏 南京 210023
甘草在中药复方中应用广泛,能够解毒、调和诸药,被称为药中“国老”。甘草活性成分在现代临床中也可与各种化学药物或天然产物单体联合用药。甘草及其活性成分的配伍用药作用机制可能与其改变药物的溶解度或体内代谢过程等生物药剂学特性有关。综合国内外相关研究表明,甘草酸在甘草体外增溶方面起主要作用;药物跨膜转运能力的改变也多与甘草酸及其苷元甘草次酸有关;而甘草黄酮类成分则在影响药物的体内代谢方面发挥了重要作用。
甘草;甘草酸;甘草次酸;黄酮类成分;药物相互作用;增溶;跨膜转运;代谢
甘草为豆科植物甘草Fisch.、胀果甘草Bat.或光果甘草L.的干燥根和根茎,具有补脾益气、清热解毒、祛痰止咳、缓急止痛、调和诸药等功效。其味甘性平,药性缓和,常作为“使药”在方中与各类寒热补泻药物同用,以协调寒热,平调升降,缓和药物烈性,减轻不良反应,并改善组方的疗效。南朝梁代陶弘景《本草经集注》言甘草“解百药毒,为九土之精,安和七十二种石,一千二百种草”,且将其“和诸药”的作用比喻为药中“国老”;明代李时珍《本草纲目》也认为,甘草“协和群品,有元老之功”,是“药中之良相”。这种解百药毒、调和诸药的作用,使甘草成为中药方剂中最常见的药物之一,甚至产生了“十方九草”的说法。据统计,《伤寒论》111首内服方剂中,有70首都使用了甘草,比例高达六成;《金匮要略》205首方中,也有超四成使用了甘草[1]。这催生了诸如甘草-芍药、桔梗-甘草、人参-甘草、茯苓-甘草等经典药对[2]。除中药间的配伍外,临床上也有将甘草活性成分与化学药物联用的报道[3-4]。现代研究表明,甘草与药物的相互作用和配伍机制主要体现在2个方面,一方面与其活性成分的药理作用有关,如甘草酸通过肾上腺素能β2受体抗哮喘的作用可与沙丁胺醇协同[5];另一方面则与生物药剂学特性有关,如联用甘草可改变芍药苷[6]、桔梗皂苷[7]、瑞香素[8]、辛伐他汀[9]等中药活性成分或化学药物的生物利用度。本文根据配伍甘草及其活性成分后药物溶解度、跨膜转运和肝脏代谢等发生的变化,从生物药剂学视角为甘草“调和诸药”的作用机制提供科学依据。
1 甘草活性成分的体内代谢过程
1.1 三萜类成分
三萜皂苷是甘草中最主要的活性部位,甘草酸是其代表。甘草酸又名甘草皂苷,在植物体内主要以盐的形式存在,被称为甘草甜素。研究表明,甘草酸的口服生物利用度仅4.0%,主要经肠道微生物水解直接或分步脱去两分子葡萄糖醛酸(glucuronic acid,GluA)生成皂苷元甘草次酸后被吸收[10]。甘草次酸进入血液后在肝脏细胞色素P450同工酶(cytochrome P450 isoenzyme,CYP450s)、UDP- GluA转移酶(UDP-glucuronyl transferases,UGTs)等代谢酶的作用下发生羟化(+OH)或GluA化(+GluA),生成相应的代谢产物[10-11]。甘草酸和甘草次酸的体内代谢过程见图1。
1.2 黄酮类成分
甘草苷和异甘草苷是甘草中最常见的黄酮苷类成分。甘草苷可经消化道微生物作用,脱葡萄糖(glucose,Glc)为苷元甘草素后被吸收入血[12],后者则被肝脏CYP450s、UGTs羟化或GluA化生成相应的代谢产物[13-14]。异甘草苷的体内过程与之类似[14],甘草苷和甘草素的体内代谢途径见图2。
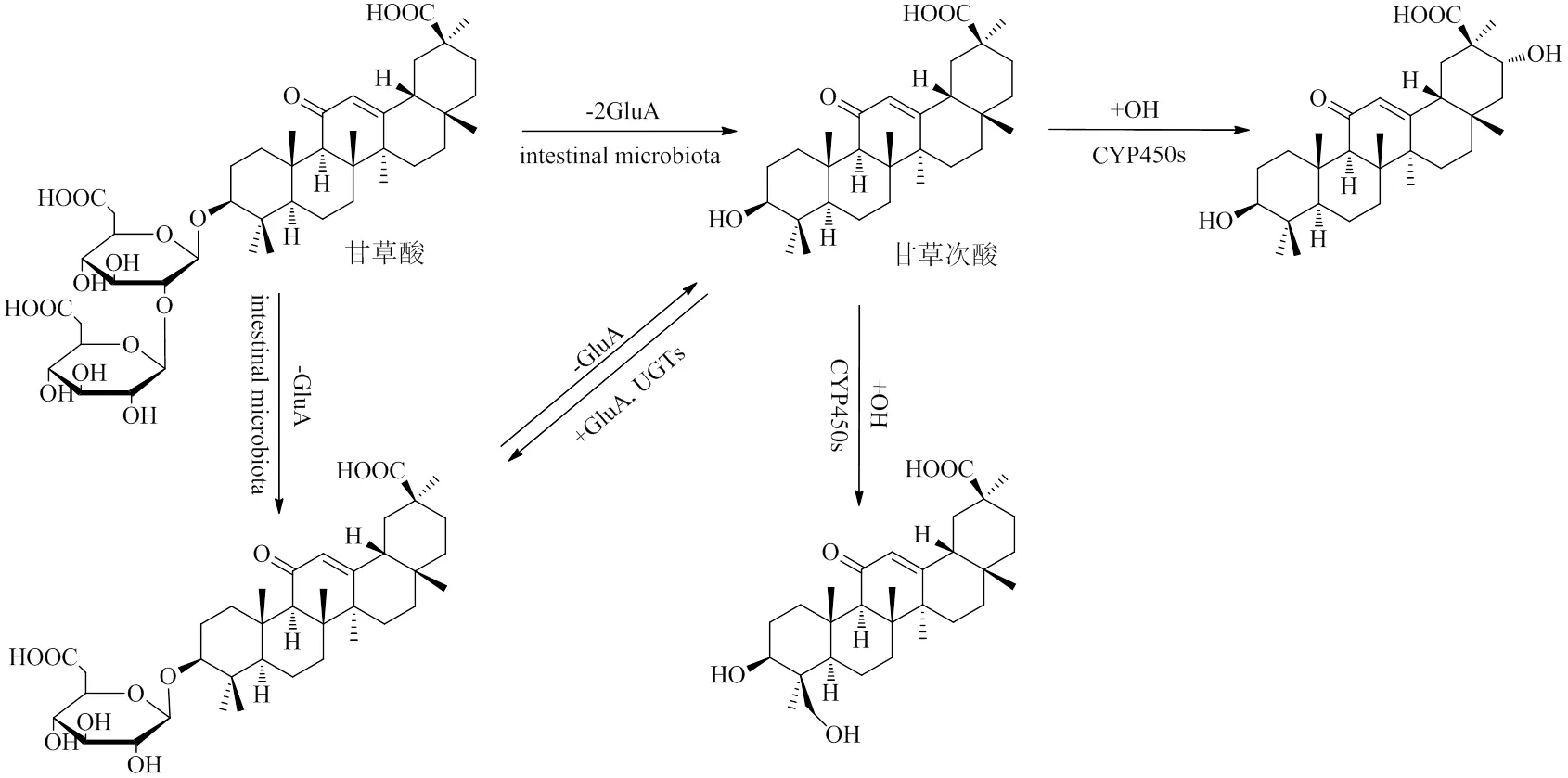
图1 甘草酸和甘草次酸的体内代谢

图2 甘草苷和甘草素的体内代谢
甘草查尔酮A是胀果甘草中一种特有的黄酮类成分,近年国内外也对其活性及体内代谢过程展开了研究,发现其被吸收入血后2个酚羟基位点可能分别发生GluA化,推测与UGTs的作用有关[15]。
2 甘草及其活性成分的增溶作用
近年来,随着中药超分子化学理论的提出和发展,中药水煎液各成分分子间的非共价键相互作用日益受到关注,煎液中一些具有特殊结构的成分分子,可能通过络合、包合等作用,以自组装等形式与其他成分分子形成超分子复合物,影响后者在煎液中的溶解性能[16]。陶叶琴等[17]基于超分子“印迹模板”理论研究了甘草的增溶特征,发现甘草对升麻葛根汤等7种中药复方汤剂有增溶作用,提示了从增溶角度认识甘草配伍用药作用机制的可能性。目前,对于甘草增溶作用的机制研究主要集中于其三萜皂苷类化学成分甘草酸的超分子自组装特性。
甘草酸分子由疏水的三萜烯和亲水的糖链2部分组成,具有两亲性,可通过疏水相互作用自组装形成非共价复合物[18]。有报道称,当甘草酸的浓度在0.01~1 mmol/L时,溶液中可以观察到甘草酸的二聚体复合物;而当浓度大于1 mmol/L时,则能形成大的胶束样聚集体[19]。低浓度条件下生成的甘草酸二聚体可将疏水分子结合于其环面内,形成“主-客复合物”以增加后者的溶解度;且这种复合物结构与环糊精的刚性固定结构相比,更利于与相对分子质量过大或过小的疏水分子结合[20]。而甘草酸胶束样聚集体则一般在高浓度条件下形成,且在中低pH值条件下稳定性高[19],Matsuoka等[21]也利用小角度X射线散射法印证了这一点,pH值为5或6时甘草酸的临界胶束浓度分别为2.9、5.3 mmol/L,而pH值大于7时则无法得到确切的临界胶束浓度。这种由两亲性分子在水中形成的胶束被认为具有亲水性的外壳和亲脂性的内核,可用于负载疏水性药物[22]。已有许多研究利用甘草酸的这种超分子自组装行为,开发出新型递药系统,提高疏水药物溶解性能的同时,也能利用甘草酸的药理特性发挥一些作用[21-27]。甘草酸胶束新剂型及性能见表1。

表1 甘草酸胶束新剂型及性能
甘草酸的超分子自组装特性可解释其对一些疏水性中药活性成分或化学药的增溶机制,并借此开发一些新型递药系统,但尚无法诠释甘草在复方煎液中的增溶机制。原因如下:(1)甘草酸在复方煎液中的浓度有限,是否能形成二聚体或更大的聚集体需要进一步证实;(2)甘草中并非只有甘草酸具有超分子自组装特性,报道称甘草蛋白也有此作用[29];(3)复方煎液中成分分子的非共价结合方式很多,中药超分子复合物的形成方式也并不限于包合,甘草活性成分在复方煎液中与其他药物成分的作用方式可能是多样化的,需深入探究。
3 甘草及其活性成分在跨膜转运方面的作用
跨膜转运影响药物的吸收、分布等体内过程。外排转运体的表达和活性、膜渗透性的强弱和细胞间紧密连接的状态是影响药物跨膜转运的重要因素。许多学者认为甘草及其活性成分可能通过作用于这些环节,改变中药组分或化学药物的跨膜转运能力,以改变药物的吸收和体内分布,从而发挥调和诸药的作用。
3.1 对外排转运体的作用
外排转运体是一类将物质从细胞内泵出细胞外的蛋白质,包括P-糖蛋白(P-gp)、多药耐药相关蛋白(multidrug resistance-associated protein,MRP)、乳腺癌耐药蛋白(breast cancer resistant protein,BCRP)等。它们可保护细胞免受一些外源性物质的侵害,但同时也导致药物的吸收不良或耐药性。一些中药活性成分和化学药物是外排转运体的底物,甘草活性成分可能通过影响外排转运体的功能(增强或抑制)或表达(上调或下调)以改变药物的跨膜转运能力[30-31]。
3.1.1 甘草次酸对外排转运体的作用 有关甘草次酸对外排转运体的作用的研究最多。Li等[32]发现,50 μmol/L 18β-甘草次酸可抑制MDR1-MDCKⅡ和Caco-2细胞中P-gp介导的特异性底物地高辛的外排,且对前者的抑制率高达83.89%,而18α-甘草次酸、甘草苷和甘草酸则无显著作用,甘草苷和甘草酸甚至一定程度上降低细胞中地高辛浓度,但无统计学差异。Nabekura等[33]也发现,18β-甘草次酸可在KB-C2细胞中抑制P-gp介导的柔红霉素和MRP1介导的钙黄绿素外排,同等浓度的甘草酸、甘草素和异甘草素则无显著作用。除P-gp外,Yoshida等[34]还发现,甘草次酸可显著抑制Sf9细胞中MRP2和LLC-PK1和BCRP介导的[3H] E217βG外排;而Chen等[35]的研究则表明,甘草次酸可抑制大鼠肝原代细胞中的MRP4和BCRP,提高抗乙肝病毒药物恩替卡韦的细胞内积累,从而增强其疗效,甘草酸则无显著作用。这些结果提示甘草次酸可能通过抑制P-gp、MRP和BCRP等外排转运体,来促进药物在胃肠道内的吸收或降低某些靶细胞的耐药性。
然而,当Hou等[36]用甘草次酸处理LS-180细胞时,却发现其显著促进P-gp介导的罗丹明123外排;而He等[37]也得到了类似的结果,且进一步发现用甘草次酸处理3、7、10 d可显著上调P-gp蛋白的mRNA表达;另外,在人肾小管上皮HK-2细胞模型中,甘草次酸也被发现可显著降低细胞中雷公藤甲素的浓度,且此作用可被P-gp抑制剂维拉帕米逆转,表明甘草次酸对P-gp介导的雷公藤甲素外排具有促进作用,提示其或可降低后者的肾毒性[38]。甘草酸对外排转运体的作用见表2。

表2 甘草次酸对外排转运体的作用
IC50-半数抑制浓度,下表同
IC50-inhibitory concentration, same as below tables
3.1.2 甘草酸对外排转运体的作用 与甘草次酸的争议性不同,甘草酸及其盐对P-gp的增强作用报道较多。Guo等[39]报道,甘草酸通过增强P-gp活性,增强Caco-2细胞对积雪草酸的外排作用,并在大鼠体内药动学实验中得到了验证。其他药物或天然产物,如葛根素[40]、扁蒴藤素[41]、雷公藤红素[42]等均有被甘草酸促进P-gp外排的报道。这些发现暗示了甘草酸或可降低一些药物的胃肠道吸收或进入肝肾细胞的能力,以减小毒性。不过也有少数报道认为甘草酸及其盐会抑制P-gp的外排作用,如甘草酸二铵对乌头碱在肠道中的吸收促进作用就被归因于此[43]。
甘草酸及其盐对其他外排转运体的作用报道则相对较少。就MRP而言,报道发现甘草酸及其盐对MRP4无显著作用[35],但作为竞争性底物一定程度上可抑制MRP2和MRP3对其他药物的外排,在逆转肝癌细胞的顺铂耐药性[44]、减少谷胱甘肽的胆汁排泄[45]等方面可能有一定贡献。甘草酸对外排转运体作用见表3。
3.1.3 甘草黄酮类成分对外排转运体的作用 研究发现,10 μmol/L异甘草苷、异甘草素、甘草苷、甘草素和甘草查尔酮A处理3、7、10 d均能显著上调P-gp、BCRP和MRP2 3种外排转运蛋白的表达,尤以异甘草苷和异甘草素对BCRP的作用最明显;且这些成分在5~25 μmol/L均显示出对LS-180细胞中罗丹明123的外排增强作用[37]。此外,光果甘草中的黄酮类成分光甘草定作为P-gp的底物,则被发现能强烈抑制Caco-2细胞中P-gp介导的地高辛外排,可能对药物的肠道吸收有一定积极意义[46]。甘草黄酮类成分对外排转运体作用见表3。

表3 甘草酸和甘草黄酮类成分对外排转运体的作用
综合国内外学者对甘草活性成分在外排转运体方面的作用研究,可以发现,开展的工作虽多,但尚难以得出系统性结论,主要存在以下问题:(1)甘草不同活性成分对外排转运体的作用不尽相同,如甘草次酸多被报道抑制外排,而甘草酸多被报道能促进外排;(2)同一活性物质的不同构型对外排转运体作用不同,如分子对接模拟和实验均表明18α-甘草次酸对P-gp的作用不如18β-甘草次酸显著[32];(3)同一活性物质对不同外排转运体的作用亦不尽相同,如甘草酸对P-gp和MRPs的作用可能相反;(4)口服的甘草提取物是这些活性成分的混合物,且这些活性成分到达靶部位后结构可能发生变化,使得甘草对药物吸收或耐药性综合影响变得更加难以预测。对此,建议综合多种模型来考察甘草对外排转运体的作用机制。一方面,不同的细胞转运模型特点各不相同,LS-180与Caco-2细胞相比具有更高的P-gp表达水平[37],适用于甘草对药物小肠吸收的作用研究;Sf9、MDCK则可建立MRP高表达的细胞模型,适用于研究甘草抗耐药作用。另一方面,体外细胞转运模型、小肠离体转运模型(肠襻法、外翻肠囊法等)、在体肠灌流模型、体内药动学模型各有优劣,体内外多模型联合更利于阐释甘草及其活性成分对药物肠道吸收的影响。此外,给药浓度、作用时间、蛋白表达量的改变均会影响体外模型结果[47],应纳入综合分析。
3.2 对细胞膜渗透性的作用
近年来,有研究开始从改善细胞膜渗透性的角度探讨甘草活性成分对药物跨膜转运的影响,其主要研究对象是甘草酸及其盐。该研究以人红细胞为模型,发现甘草酸盐可以降低细胞膜的弹性模量并增加甲酸钠的膜渗透性,提示其对细胞膜弹性和通透性可能有提高作用[48]。进一步选择人工双层脂质分子膜二棕榈酰磷脂酰胆碱(dipalmitoyl phosphatidylcholine,DPPC)、棕榈酰油酰磷脂酰胆碱和二油酰磷脂酰胆碱为模型,使用动态NMR和分子动力学技术,考察甘草酸对脂质双分子层的影响,结果发现90%甘草酸会沉降于膜表面,其中80%又能嵌入脂质双分子层,并长期停留于脂质双层的外半层亲水头部和疏水尾部之间,甚至在刚性最强的DPPC膜中可以到达内半层,使膜变薄,通透性增强[49]。这一发现或能解释甘草酸提高细胞膜渗透性的作用机制,且已被用来解释甘草酸促进甲酸钠的红细胞渗透[48]、辛伐他汀的胃肠道吸收[9]等现象。研究显示,作为一种以被动扩散方式跨膜转运的药物,驱虫药吡喹酮与甘草酸二钠配伍后在人工膜和单层Caco-2细胞上的渗透速率均显著提高,证实了甘草酸及其盐的促渗作用[50]。考虑到甘草酸能增加疏水中药成分或化学药分子的溶解度,将促进细胞膜渗透与增加体外溶解度结合起来,或可更全面的解释甘草与疏水药物的配伍用药机制[51]。
3.3 对细胞旁路途径的作用
细胞旁路途径转运也是药物跨膜转运的方式之一,药物分子直接通过上皮细胞之间的紧密连接进入细胞间隙,从而跨过生物膜。细胞旁路途径吸收主要受细胞间紧密连接调控,一些口服吸收促进剂可打开紧密连接,促进药物的胃肠道吸收。研究表明,甘草的活性成分或有一定调控胃肠道细胞紧密连接的作用。Imai等[52]以甘草酸二钾为对象展开了研究,发现单独给予甘草酸二钾不能降低Caco-2细胞的跨膜电阻值,而与另一种吸收促进剂癸酸钠联用则能快速且持久的降低跨膜电阻值,打开细胞间紧密连接,促进降钙素的结肠吸收。而18β-甘草次酸则被报道能促进肝素的肠道吸收,具体机制尚不明确,推测与细胞旁路途径有关[53-54]。
4 甘草及其活性成分在肝脏代谢方面作用
肝脏是药物代谢的主要器官,药物在肝脏内主要有Ⅰ相反应和Ⅱ相反应,前者使药物分子发生氧化、水解或异构化,极性增大,水溶性增强,易于排泄,又称官能团反应;后者则可使药物及其Ⅰ相代谢产物与一些内源性物质(如GluA)结合,进一步增强水溶性,因而又称结合反应。肝脏对药物的代谢可使药物失活或活性增强,也可能将药物转化为一些对机体有害的代谢物。传统认为甘草有解百药毒的作用,其极有可能通过诱导肝脏的代谢以降低某些有毒药物的毒性,或抑制肝脏的代谢以减少某些药物生成的肝毒性亲电子代谢物。因此已有相当多的研究致力于从肝脏代谢角度解释甘草及其活性成分的配伍用药机制。
4.1 对CYP450s的作用
CYP450s是催化一相代谢的关键酶,其主要有CYP1、2、3这3个家族,均在药物的肝脏代谢中扮演了重要的角色[55],见表4。甘草及其活性成分可能通过诱导或抑制CYP450s以影响其他药物或中药成分的体内代谢。

表4 参与药物代谢的代表性CYP450s及其代谢作用
4.1.1 甘草酸和甘草次酸对CYP450s的作用 甘草酸对CYP1和CYP2亚家族的作用不是很显著[56],但对CYP3亚家族有显著抑制作用。甘草酸可在50~500 μmol/L剂量相关性活化孕烷X受体以提高HepG2细胞中CYP3A4的蛋白表达[57],表明甘草酸对CYP3A4具有一定的诱导作用;而每日ig大鼠100 mg/kg甘草酸也能显著增加CYP3A4的体内代谢能力,这或可解释甘草酸促进雷公藤内酯代谢的作用[58]。然而如前述,大多数甘草酸须在结肠内被肠道微生物代谢为甘草次酸后被吸收,许多研究者将目光转向了甘草次酸。
研究人员使用人肝微粒对甘草次酸和非那西汀进行体外共孵育,发现甘草次酸对CYP1A2介导的非那西汀脱乙基反应有一定抑制作用,但IC50远高于特异性抑制剂α-萘黄酮的1.35 μmol/L,作用微弱[59-60]。且其对CYP2A6[61]、CYP2D6[60-62]和CYP2C8[59,62]的抑制作用均不是很显著,但抑制CYP2C19的作用较好[59]。甘草次酸不能抑制CYP2E1活性[59],但可下调其在小鼠肝脏的表达[63-64],提示了降低氯仿和对乙酰氨基酚肝毒性的潜力。此外,针对CYP3亚家族,研究表明25 μmol/L甘草次酸可显著增加CYP3A4的活性[36];Chen等[65]也发现甘草次酸可上调CYP3A4的mRNA表达,这与前述甘草酸的作用基本一致。然而亦有许多学者却在大鼠肝微粒孵育实验中发现甘草次酸对CYP3A4介导的咪达唑仑羟化有抑制作用[59-60,62,66];同时一些体内实验也印证了这一点[61]。这种矛盾的结果可能与实验设计有关,需要进一步研究探讨[47]。
综上所述,甘草酸主要上调和诱导CYP3A4,而甘草次酸则抑制CYP2C19和CYP2E1,对CYP3A4的作用存在一定争议。甘草酸和甘草次酸对CYP450s的作用见表5。
4.1.2 甘草黄酮对CYP450s的作用 作为甘草中比较常见的黄酮类成分,甘草素和异甘草素被报道在HepG2细胞中对多种CYP450的mRNA表达有调节作用,其中对CYP1A2 mRNA表达的诱导作用最显著[65]。另有研究则从甘草提取物入手[67],发现光果甘草和乌拉尔甘草根的提取物对CYP1A2抑制作用较弱,而相比之下胀果甘草则作用较强,原因在于后者具有甘草查尔酮A这一独有的黄酮类成分,而该成分是CYP1A2的混合型抑制剂,IC50仅为1.15 μmol/L[68],效果十分显著。此外,该物质对CYP2D6、2C8、2C9、2C19和3A4均有显著抑制作用[65,68],并可降低CYP2C19、2D6和3A4的mRNA表达[65],但对CYP2E1作用较弱[67]。
综上所述,CYP1A2的活性和表达主要受甘草中黄酮类成分调控,且其中的甘草查尔酮A还对CYP3A4和除2E1以外的CYP2家族具有强烈抑制作用。由于黄芩素、大黄素和蛇床子素等中药成分为CYP2C亚家族酶的底物[69],甘草极有可能通过抑制这些成分的肝脏代谢以减少其消除,延长其体内停留时间以增加疗效。甘草黄酮对CYP450s的作用见表5。
4.2 对UGTs的作用
UGTs是一类重要的催化二相代谢的酶,可使一些带有羟基、氨基或羧基的药物或一相代谢物发生GluA化反应,生成水溶性更高的代谢物,便于排泄。研究发现,大鼠长期摄入甘草次酸可诱导肝脏UGTs mRNA的表达[70],而在体外肝微粒孵育实验中,甘草次酸则显示出对UGTs活性显著的抑制作用[71-73]。这表明,甘草次酸虽然可以诱导UGTs的表达,但其本身也是UGTs的底物,因而可能一定程度上抑制UGTs的活性[47]。此外,甘草查尔酮A[74]、甘草素[75]等黄酮类成分均被证实为UGTs有效抑制剂。
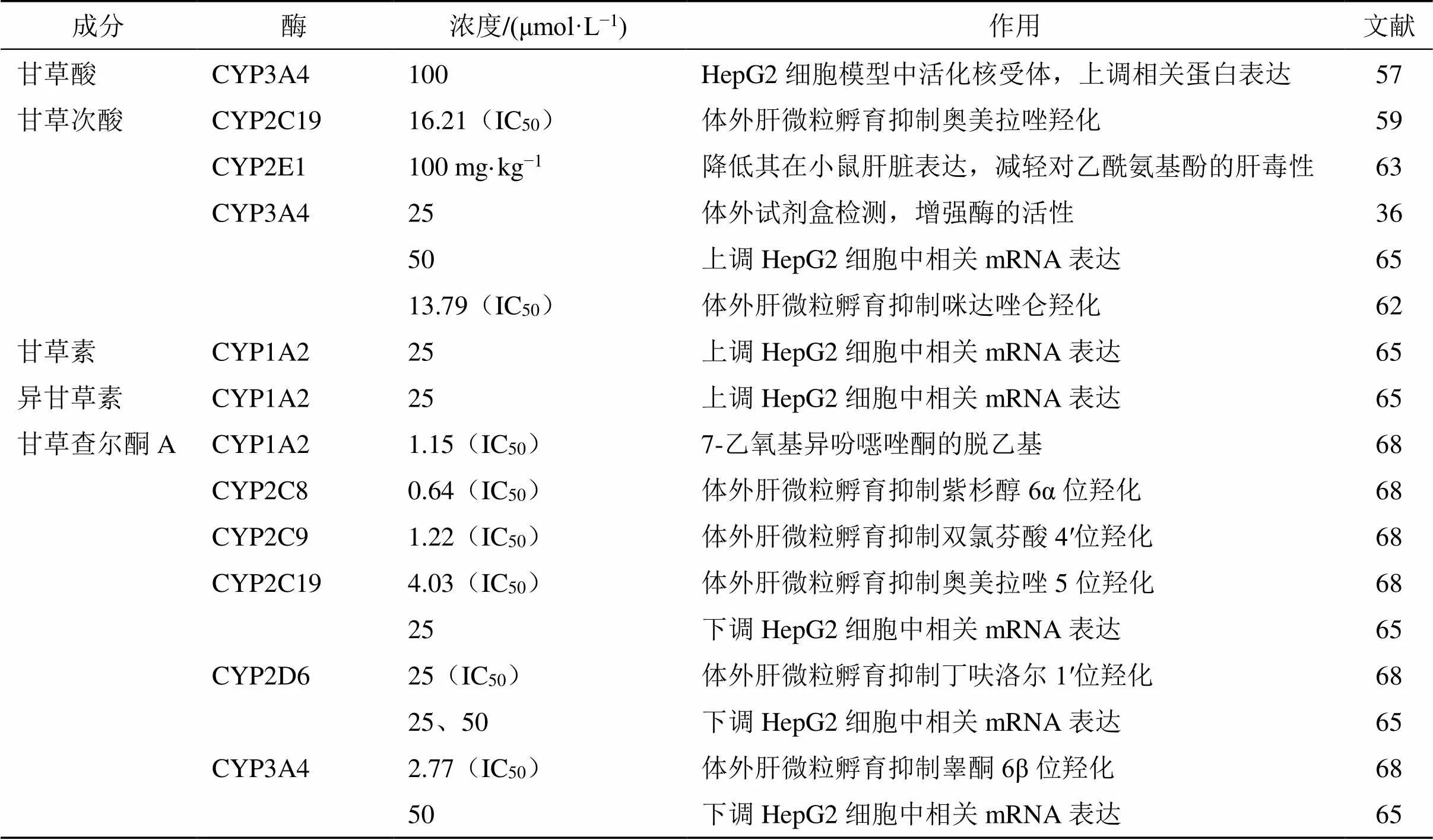
表5 甘草活性成分对CYP450s的作用
5 结语
综合国内外学者对甘草及其活性成分在生物药剂学方面的配伍用药机制研究可以发现,甘草酸及其盐在改善药物的体外溶解度和细胞膜渗透性方面起到了主要作用,可能促进药物的溶解和渗透,并具有作为载体辅料制备疏水性药物新型递药系统的潜力;甘草次酸主要下调肝脏CYP2E1,起到保肝作用,而对细胞外排转运体和肝脏CYP3A4的作用存在一定争议;黄酮类成分甘草苷、异甘草苷、甘草素和异甘草素上调细胞外排转运体和肝脏CYP1A2的表达,起到“解毒”作用;光甘草定抑制小肠对药物的外排;甘草查尔酮A则对CYP1A2、CYP2C亚家族和CYP3A4有强烈抑制作用,可能抑制一些中药活性成分的体内消除。
目前,大多数研究还是针对甘草活性成分的作用展开,这或可揭示甘草单体活性成分与一些化学药的联用机制,但想完全解释甘草在复方中的配伍机制还存在一定困难;甚至这些活性成分的作用机制本身还存在争议,尚未达成共识。基于以上困难,认为:(1)应开展更高效系统的筛选工作,借助一些高通量筛选手段,考察甘草活性成分对药物转运体和肝脏代谢酶等的作用;(2)在研究中要综合各种实验方法和模型,得出更全面、深入的结果。如研究甘草次酸对P-gp的作用时,既要考虑其对蛋白活性的影响,也要研究其对蛋白表达的作用;而研究其对蛋白活性影响时,也要综合细胞、体外组织、体内等多种模型;(3)鉴于苷类化合物多经肠道菌群水解成为苷元后被吸收,同样作为水解底物的甘草酸和甘草苷等物质是否在这一层面干预了药物的体内过程,尚缺乏研究,值得进一步探索。
利益冲突 所有作者均声明不存在利益冲突
[1] 王骞, 龚学忠. 从仲景方看甘草的临床应用 [J]. 世界中西医结合杂志, 2013, 8(4): 327-329.
[2] 曹玉洁, 唐于平, 沈娟, 等. 基于数据挖掘分析甘草药对配伍应用规律 [J]. 中草药, 2017, 48(21): 4552-4559.
[3] 萍俞, 徒康宛, 严志涵, 等. 异甘草酸镁联合恩替卡韦对慢性乙型肝炎HBeAg阳性患者治疗指标的影响研究 [J]. 中华医院感染学杂志, 2015, 25(20): 4688-4690.
[4] 张鸫媛, 任万明, 石春蕊, 等. 复方甘草酸苷与阿维A胶囊联合治疗银屑病疗效和安全性的系统评价 [J]. 中国循证医学杂志, 2013, 13(1): 112-120.
[5] Yang Y, Shi Q, Liu Z,. The synergistic anti-asthmatic effects of glycyrrhizin and salbutamol [J]., 2010, 31(4): 443-449.
[6] 刘卉, 单进军, 康安, 等. 甘草酸和甘草次酸对芍药苷在大鼠体内药动学参数的影响 [J]. 中草药, 2013, 44(12): 1610-1614.
[7] Shan J J, Zou J S, Xie T,. Effects of Gancao on pharmacokinetic profiles of platycodin D and deapio- platycodin D in Jiegeng [J]., 2015, 170: 50-56.
[8] Zhang W, Di L Q, Li J S,. The effects ofand its major bioactive components on pharmacokinetics of daphnetin inin rats [J]., 2014, 154(3): 584-592.
[9] Kong R P, Zhu X Y, Meteleva E S,. Enhanced solubility and bioavailability of simvastatin by mechanochemically obtained complexes [J]., 2017, 534(1/2): 108-118.
[10] 宋玮, 郑伟, 张洁, 等. 中药皂苷类成分的体内代谢研究进展 [J]. 药学学报, 2018, 53(10): 1609-1619.
[11] Koyama M, Shirahata T, Hirashima R,. Inhibition of UDP-glucuronosyltransferase (UGT)-mediated glycyrrhetinicacid 3--glucuronidation by polyphenols and triterpenoids [J]., 2017, 32(4): 218-223.
[12] 董世奇, 樊慧蓉, 李全胜, 等. 甘草苷在大鼠体内的代谢途径研究 [J]. 中草药, 2014, 45(17): 2499-2505.
[13] Wang A X, Hu Y, Liu H X,. C5-Hydroxylation of liquiritigenin is catalyzed selectively by CYP1A2 [J]., 2011, 41(5): 349-357.
[14] Xiang C, Qiao X, Wang Q,. From single compounds to herbal extract: A strategy to systematically characterize the metabolites of licorice in rats [J]., 2011, 39(9): 1597-1608.
[15] 杨琳. 四种黄酮类成分的血中代谢产物研究 [D]. 沈阳: 沈阳药科大学, 2007.
[16] 贺福元, 周逸群, 邓凯文, 等. 超分子化学对中医药理论的特殊影响 [J]. 中国中药杂志, 2014, 39(8): 1534- 1543.
[17] 陶叶琴, 唐闻汉, 刘金玲, 等. 基于超分子“印迹模板”理论的甘草增助溶特征研究 [J]. 中国中药杂志, 2016, 41(10): 1849-1854.
[18] Zelikman M V, Kim A V, Medvedev N N,. Structure of dimers of glycyrrhizic acid in water and their complexes with cholesterol: Molecular dynamics simulation [J]., 2015, 56(1): 67-76.
[19] Polyakov N E. Glycyrrhizic acid as a novel drug delivery vector: Synergy of drug transport and efficacy [J]., 2011, 2(1): 64-72.
[20] Polyakov N E, Leshina T V, Salakhutdinov N F,. Host-guest complexes of carotenoids with beta-glycyrrhizic acid [J]., 2006, 110(13): 6991-6998.
[21] Matsuoka K, Miyajima R, Ishida Y,. Aggregate formation of glycyrrhizic acid [J]., 2016, 500: 112-117.
[22] Nagarajan R. Constructing a molecular theory of self-assembly: Interplay of ideas from surfactants and block copolymers [J]., 2017, 244: 113-123.
[23] Wang Y T, Zhao B X, Wang S Q,. Formulation and evaluation of novel glycyrrhizic acid micelles for transdermal delivery of podophyllotoxin [J]., 2016, 23(5): 1623-1635.
[24] Zhang Q H, Polyakov N E, Chistyachenko Y S,. Preparation of curcumin self-micelle solid dispersion with enhanced bioavailability and cytotoxic activity by mechanochemistry [J]., 2018, 25(1): 198-209.
[25] Yang F H, Zhang Q, Liang Q Y,. Bioavailability enhancement of paclitaxel via a novel oral drug delivery system: Paclitaxel-loaded glycyrrhizic acid micelles [J]., 2015, 20(3): 4337-4356.
[26] Polyakov N E, Khan V K, Taraban M B,. Complexation of lappaconitine with glycyrrhizic acid: Stability and reactivity studies [J]., 2005, 109(51): 24526-24530.
[27] Polyakov N E, Khan V K, Taraban M B,. Complex of calcium receptor blocker nifedipine with glycyrrhizic acid [J]., 2008, 112(14): 4435-4440.
[28] Apanasenko I E, Selyutina O Y, Polyakov N E,. Solubilization and stabilization of macular carotenoids by water soluble oligosaccharides and polysaccharides [J]., 2015, 572: 58-65.
[29] 李冰洁, 沈勇, 廖日滔, 等. 从蛋白质自组装的角度探析甘草附子配伍减毒机制 [J]. 中国中药杂志, 2015, 40(04): 661-666.
[30] 彭燕, 谭晓斌, 贾晓斌. 甘草总皂苷及甘草酸对Caco-2细胞P-gp功能和表达的影响 [J]. 中成药, 2013, 35(9): 1846-1851.
[31] 彭燕, 谭晓斌, 贾晓斌. 甘草黄酮类成分对Caco-2细胞P-糖蛋白功能和表达的影响 [J]. 中草药, 2013, 44(19): 2703-2709.
[32] Li X, Hu J P, Wang B L,. Inhibitory effects of herbal constituents on P-glycoproteinand: Herb-drug interactions mediated via P-gp [J]., 2014, 275(2): 163-175.
[33] Nabekura T, Yamaki T, Ueno K,. Inhibition of P-glycoprotein and multidrug resistance protein 1 by dietary phytochemicals [J]., 2008, 62(5): 867-873.
[34] Yoshida N, Takada T, Yamamura Y,. Inhibitory effects of terpenoids on multidrug resistance-associated protein 2- and breast cancer resistance protein-mediated transport [J]., 2008, 36(7): 1206-1211.
[35] Chen Q Y, Chen H Z, Wang W J,. Glycyrrhetic acid, but not glycyrrhizic acid, strengthened entecavir activity by promoting its subcellular distribution in the liver via efflux inhibition [J]., 2017, 106: 313- 327.
[36] Hou Y C, Lin S P, Chao P D L. Liquorice reduced cyclosporine bioavailability by activating P-glycoprotein and CYP 3A [J]., 2012, 135(4): 2307-2312.
[37] He Y F, Ci X Y, Xie Y,. Potential detoxification effect of active ingredients in liquorice by upregulating efflux transporter [J]., 2019, 56: 175-182.
[38] Li Z H, Yan M, Cao L J,. Glycyrrhetinic acid accelerates the clearance of triptolide through P-gp[J]., 2017, 31(7): 1090-1096.
[39] Guo L, Cui Y, Hao K J. Effects of glycyrrhizin on the pharmacokinetics of Asiatic acid in rats and its potential mechanism [J]., 2018, 56(1): 119-123.
[40] Zhao Q, Wang Y L, Wang H Q,. Effects of glycyrrhizin on the pharmacokinetics of puerarin in rats [J]., 2018, 48(11): 1157-1163.
[41] Zhao X F, Wu Y, Wang D M. Effects of glycyrrhizic acid on the pharmacokinetics of pristimerin in rats and its potential mechanism [J]., 2018, 43(1): 63-68.
[42] Yan G K, Zhang H H, Wang W,. Investigation of the influence of glycyrrhizin on the pharmacokinetics of celastrol in rats using LC-MS and its potential mechanism [J]., 2017, 47(7): 607-613.
[43] Chen L, Yang J, Davey A K,. Effects of diammonium glycyrrhizinate on the pharmacokinetics of aconitine in rats and the potential mechanism [J]., 2009, 39(12): 955-963.
[44] Wakamatsu T, Nakahashi Y, Hachimine D,. The combination of glycyrrhizin and lamivudine can reverse the cisplatin resistance in hepatocellular carcinoma cells through inhibition of multidrug resistance-associated proteins [J]., 2007: 31(6): 1465-1472.
[45] Xu R J, Zhang X Y, Yang J,. Effects of glycyrrhizin on biliary transport and hepatic levels of glutathione in rats [J]., 2012, 33(5): 235-245.
[46] Cao J, Chen X, Liang J,. Role of P-glycoprotein in the intestinal absorption of glabridin, an active flavonoid from the root of[J]., 2007, 35(4): 539-553.
[47] Feng X C, Ding L Q, Qiu F. Potential drug interactions associated with glycyrrhizin and glycyrrhetinic acid [J]., 2015, 47(2): 229-238.
[48] Selyutina O Y, Polyakov N E, Korneev D V,. Influence of glycyrrhizin on permeability and elasticity of cell membrane: Perspectives for drugs delivery [J]., 2016, 23(3): 848-855.
[49] Selyutina O Y, Apanasenko I E, Kim A V,. Spectroscopic and molecular dynamics characterization of glycyrrhizin membrane-modifying activity [J]., 2016, 147: 459-466.
[50] Meteleva E S, Chistyachenko Y S, Suntsova L P,. Disodium salt of glycyrrhizic acid - A novel supramolecular delivery system for anthelmintic drug praziquantel [J]., 2019, 50: 66-77.
[51] Kim A V, Shelepova E A, Selyutina O Y,. Glycyrrhizin-assisted transport of praziquantel anthelmintic drug through the lipid membrane: An experiment and MD simulation [J]., 2019, 16(7): 3188-3198.
[52] Imai T, Sakai M, Ohtake H,. Absorption-enhancing effect of glycyrrhizin induced in the presence of capric acid [J]., 2005, 294(1/2): 11-21.
[53] Motlekar N A, Srivenugopal K S, Wachtel M S,. Evaluation of the oral bioavailability of low molecular weight heparin formulated with glycyrrhetinic acid as permeation enhancer [J]., 2006, 67(2): 166-174.
[54] Neves A R, Correia-Da-silva M, Sousa E,. Strategies to overcome heparins’ low oral bioavailability [J].(Basel), 2016, 9(3): 37.
[55] Li G N, Huang K, Nikolic D,. High-throughput cytochrome P450 cocktail inhibition assay for assessing drug-drug and drug-botanical interactions [J]., 2015, 43(11): 1670-1678.
[56] Pandit S, Ponnusankar S, Bandyopadhyay A,. Exploring the possible metabolism mediated interaction ofextract with CYP3A4 and CYP2D6 [J]., 2011, 25(10): 1429-1434.
[57] Wang Y G, Zhou J M, Ma Z C,. Pregnane X receptor mediated-transcription regulation of CYP3A by glycyrrhizin: A possible mechanism for its hepatoprotective property against lithocholic acid-induced injury [J]., 2012, 200(1): 11-20.
[58] Tai T, Huang X, Su Y W,. Glycyrrhizin accelerates the metabolism of triptolide through induction of CYP3A in rats [J]., 2014, 152(2): 358-363.
[59] 刘丽, 肖娟, 彭志红, 等. 甘草次酸在人细胞色素CYP450中体外代谢研究(英文) [J]. 药学学报, 2011, 46(1): 81-87.
[60] Li A F, Ma N N, Zhao Z J,. Glycyrrhetinic acid might increase the nephrotoxicity of bakuchiol by inhibiting cytochrome P450 isoenzymes [J]., 2016, 4: e2723.
[61] Lv Q L, Wang G H, Chen S H,.andinhibitory effects of glycyrrhetinic acid in mice and human cytochrome P450 3A4 [J]., 2015, 13(1): 84.
[62] Zhao K, Ding M, Cao H,.metabolism of glycyrrhetinic acid by human and rat liver microsomes and its interactions with six CYP substrates [J]., 2012, 64(10): 1445-1451.
[63] Yang G L, Zhang L, Ma L,. Glycyrrhetinic acid prevents acetaminophen-induced acute liver injury via the inhibition of CYP2E1 expression and HMGB1-TLR4 signal activation in mice [J]., 2017, 50: 186-193.
[64] Jeong H G, You H J, Park S J,. Hepatoprotective effects of 18beta-glycyrrhetinic acid on carbon tetrachloride- induced liver injury: inhibition of cytochrome P450 2E1 expression [J]., 2002, 46(3): 221-227.
[65] Chen H, Zhang X M, Feng Y F,. Bioactive components ofmediate drug functions and properties through regulation of CYP450 enzymes [J]., 2014, 10(3): 1355-1362.
[66] Li H Y, Xu W, Su J,.andinhibitory effects of glycyrrhetinic acid on cytochrome P450 3A activity [J]., 2010, 86(5/6): 287-292.
[67] Li G N, Simmler C, Chen L Y,. Cytochrome P450 inhibition by three licorice species and fourteen licorice constituents [J]., 2017, 109: 182-190.
[68] He W, Wu J J, Ning J,. Inhibition of human cytochrome P450 enzymes by licochalcone A, a naturally occurring constituent of licorice [J]., 2015, 29(7): 1569-1576.
[69] 张远冬, 刘学庆, 郭延垒, 等. 大鼠肝微粒体法评价20种中药有效成分对CYP2C9酶的作用 [J]. 第三军医大学学报, 2013, 35(24): 2654-2658.
[70] Lee K W, Ho W S. 18β-glycyrrhetinic acid induces UDP-glucuronosyltransferase in rats [J]., 2013, 20(12): 1360-1364.
[71] Katoh M, Yoshioka Y, Nakagawa N,. Effects of Japanese herbal medicine, kampo, on human UGT1A1 activity [J]., 2009, 24(3): 226-234.
[72] Huang Y P, Cao Y F, Fang Z Z,. Glycyrrhetinic acid exhibits strong inhibitory effects towards UDP- glucuronosyltransferase (UGT) 1A3 and 2B7 [J]., 2013, 27(9): 1358-1361.
[73] Nakagawa N, Katoh M, Yoshioka Y,. Inhibitory effects of kampo medicine on human UGT2B7 activity [J]., 2009, 24(6): 490-499.
[74] Xin H, Qi X Y, Wu J J,. Assessment of the inhibition potential of licochalcone A against human UDP- glucuronosyltransferases [J]., 2016, 90: 112-122.
[75] Guo B, Fan X R, Fang Z Z,. Deglycosylation of liquiritin strongly enhances its inhibitory potential towards UDP-glucuronosyltransferase (UGT) isoforms [J]., 2013, 27(8): 1232-1236.
Research progress on biopharmaceutical mechanism of“moderating property of herbs”
LUO Zi-chen1, 2, 3, ZHANG Wen1, 2, YANG Rui3, SHAN Jin-jun3, DI Liu-qing1, 2
1. School of Pharmacy, Nanjing University of Chinese Medicine, Nanjing 210023, China 2. Jiangsu Engineering Research Center for Efficient Delivery System of TCM, Nanjing University of Chinese Medicine, Nanjing 210023, China 3. Jiangsu Key Laboratory of Pediatric Respiratory Disease (TCM), Nanjing University of Chinese Medicine, Nanjing 210023, China
, known as “Guo lao” in Chinese materia medica, is widely used in Chinese herbal compounds, which has the effect of reducing drug toxicity and improving drug efficacy. Active ingredients ofare often used in combination with various chemical drugs or natural product phytochemicals in modern clinical practice. The mechanism of interactions betweenand other drugs may be related to the change of the biopharmaceutical process of drug, such as solubility and ADME. The results of various studies show that glycyrrhizic acid plays a major role in solubilizing drugs, while changes in the membrane transport capacity of drugs are mostly related to glycyrrhizic acid and glycyrrhetic acid. Moreover, flavonoids play major role in changing the liver metabolism of drugs.
Fisch.; glycyrrhizic acid; glycyrrhetic acid; flavonoids; drug-drug interactions; solubilization; membrane transport; metabolism
R285.62
A
0253 - 2670(2021)01 - 0267 - 11
10.7501/j.issn.0253-2670.2021.01.032
2020-03-26
国家自然科学基金资助项目(81774156);国家自然科学基金资助项目(81273655);国家自然科学基金资助项目(81001499);江苏省“六大人才高峰”高层次人才选拔培养资助项目(YY-022);江苏省研究生科研与实践创新计划(SJKY19_1457)
罗子宸(1996—),男,硕士研究生,研究方向为中药代谢组学和生物药剂学。E-mail: 20181398@njucm.edu.cn
单进军(1979—),男,教授,研究方向为代谢组学与中医药。Email: dfsjj@163.com
狄留庆(1964—),男,教授,研究方向为中药药剂学。Email: diliuqing928@163.com
[责任编辑 崔艳丽]
