利用环境DNA-宏条形码技术监测苏州地区小管福寿螺的入侵
2021-01-04陈晓方靖怡王萌沈晴孙振军王备新
陈晓 方靖怡 王萌 沈晴 孙振军 王备新






摘要 :小管福寿螺Pomacea canaliculata是我国南方重要的外来入侵有害生物,目前已在江苏苏州的河道、湖塘等水体中广泛发生,并呈现向北入侵趋势。及时准确地监测小管福寿螺在水体中的入侵扩散是有效防控其为害的关键。传统观察法受其生活史、发生状况及环境等影响,难以在入侵早期及时监测。新兴的环境DNA-宏条形码(environmental DNA metabarcoding)技术可实现对入侵生物快速、灵敏的监测。本文分别采用环境DNA-宏条形码技术和传统观察方法对苏州地区河流、湖泊和运河共38个样点的小管福寿螺发生情况进行了检测。结果显示,环境DNA-宏条形码技术检测到小管福寿螺的发生率(92.11%)远高于传统观察法(36.84%)。丰度阈值的设定和水体类型对环境DNA-宏条形码技术的检测结果有一定影响,本研究为今后运用环境DNA-宏条形码技术进行福寿螺入侵监测提供技术支持。
关键词 :环境DNA(eDNA); 高通量测序; 小管福寿螺; 生物入侵
中图分类号:
S 40
文献标识码: A
DOI: 10.16688/j.zwbh.2020389
Monitoring of the invasive species Pomacea canaliculata via environmental DNA metabarcoding in Suzhou city
CHEN Xiao1, FANG Jingyi1,2, WANG Meng1, SHEN Qing3, SUN Zhenjun3*, WANG Beixin1*
(1. College of Plant Protection, Nanjing Agricultural University, Nanjing 210095, China;
2. Wageningen University, Wageningen 6700 AK, The Netherlands; 3. Suzhou
Station of Plant Protection and Plant Quarantine, Suzhou 215008, China)
Abstract
Pomacea canaliculata is a dangerous invasive species in southern China. At present, P.canaliculata has widely occurred in rivers, lakes, and other water bodies in Suzhou, Jiangsu province, and shows a trend of northward invasion. Timely and accurate monitoring of its invasion and spreading in water bodies is the key to effectively prevent and control its harm. Owing to the effect of its life history, occurrence, and environment, traditional observation cannot monitor P.canaliculata in the early stage of invasion. The emerging environmental DNA metabarcoding technology shows rapid and sensitive monitoring of invasive organisms. In this paper, environmental DNA metabarcoding and traditional observation methods were used to detect the occurrence of P.canaliculata in 38 samples of rivers, lakes, and canals in Suzhou. The results showed that the detection rate of P.canaliculata by environmental DNA metabarcoding technique (92.11%) was much higher than that by traditional observation method (36.84%). The setting of abundance threshold and water body type have a certain impact on the detection results by environmental DNA metabarcoding technology. This study would provide technical support for the future use of this technology to monitor Pomacea.
Key words
environmental DNA; high-throughput sequencing; Pomacea canaliculata; biological invasion
外來物种入侵是重要的全球环境问题和生态安全问题[1]。入侵生物对农、林、牧、渔业和交通航运等产生直接或间接的破坏,造成重大的经济损失和生态安全问题。福寿螺Pomacea属软体动物门Mollusca,腹足纲Gastropoda,瓶螺科Ampullariidae,是世界100种恶性外来入侵物种之一[2],具有适应性强、生长快、繁殖力高,食性杂且食量大等特点,在局部区域对水稻、茭白、莲藕、芡实等农作物造成严重危害。此外,福寿螺还是一些危害人体健康的寄生虫的中间宿主,如卷棘口吸虫Echinostoma revolutum和广州管圆线虫Angiostrongylus cantonensis等[3]。长期以来福寿螺被认为只有1种,即小管福寿螺Pomacea canaliculata,福寿螺通常也特指小管福寿螺,但现在通过分子生物学方法发现福寿螺属包括多个种[4]。小管福寿螺自1981年引入广东后,逐渐扩散至浙江、江西、云南和四川等地,现已成为影响南方农业生产的重要有害生物[5]。2003年中国国家环保总局将其列入首批16种外来入侵生物名单[6]。当前,小管福寿螺作为外来物种已在江苏苏州定殖[7],在部分河道、湖塘发生量较大,并呈现逐渐向北(无锡、常州、镇江、南京)扩散的趋势。入侵初期是防治入侵物种的重要时期,监测预警是有效防控生物入侵的重要手段[1]。然而,小管福寿螺生活于水中,可潜藏在水底,非繁殖期有较强的隐蔽性[6],传统的现场观察和调查问卷法发现难度大,不利于早期预警。如何有效地开展早期监测是当前福寿螺防控中的重要工作内容。
环境DNA(environmental DNA, eDNA)是指生物体经由体表、尿液、粪便、黏液或细胞死亡裂解后等释放到环境中的DNA片段[8]。随着高通量测序技术的发展,DNA宏条形码技术(DNA metabarcoding)可以从混合样本中同时获得更多的生物信息。环境DNA-宏条形码技术,即利用宏条形码技术对环境样本中分离的DNA进行扩增测序,可以同时鉴定混合样本中多个分类单元[9](后文简称为环境DNA技术)。目前,环境DNA技术已成功应用于入侵水生生物的监测,并展现出快速、灵敏等优点。该技术可以在亚洲鲤鱼入侵北美五大湖早期密度较低时检测到其存在[1011]。Takahara等[12]发现环境DNA技术相比传统观察法能更准确地反映入侵物种蓝鳃太阳鱼Lepomis macrochirus在日本的扩散趋势。此外,对云南梯田克氏原螯虾Procambarus clarkia的环境DNA监测结果也表明该技术的检出率明显高于传统的诱捕法[13]。然而,当前我国多数地区对福寿螺的发生监测依然以走访调查、现场观察等为主[1416],尚未见利用环境DNA-宏条形码技术进行监测的相关研究。入侵生物的早期预警需要在其丰度很低时监测到其存在,因此低丰度序列的阈值筛选尤为重要。研究发现,环境DNA在不同类型的水体中的富集也存在差异[17]。为此,还需明确筛选出的丰度阈值及各种环境因素对环境DNA技术监测結果的影响。
本文旨在利用环境DNA技术监测苏州地区不同水体类型的38个样点的小管福寿螺发生情况,探索环境DNA技术是否可以作为批量检测入侵生物福寿螺的灵敏、高效的技术,明确丰度阈值和水体类型对环境DNA技术检测结果的影响,为今后运用环境DNA-宏条形码技术对福寿螺进行监测和早期预警提供技术参考。
1 材料与方法
1.1 研究区域和样点设置
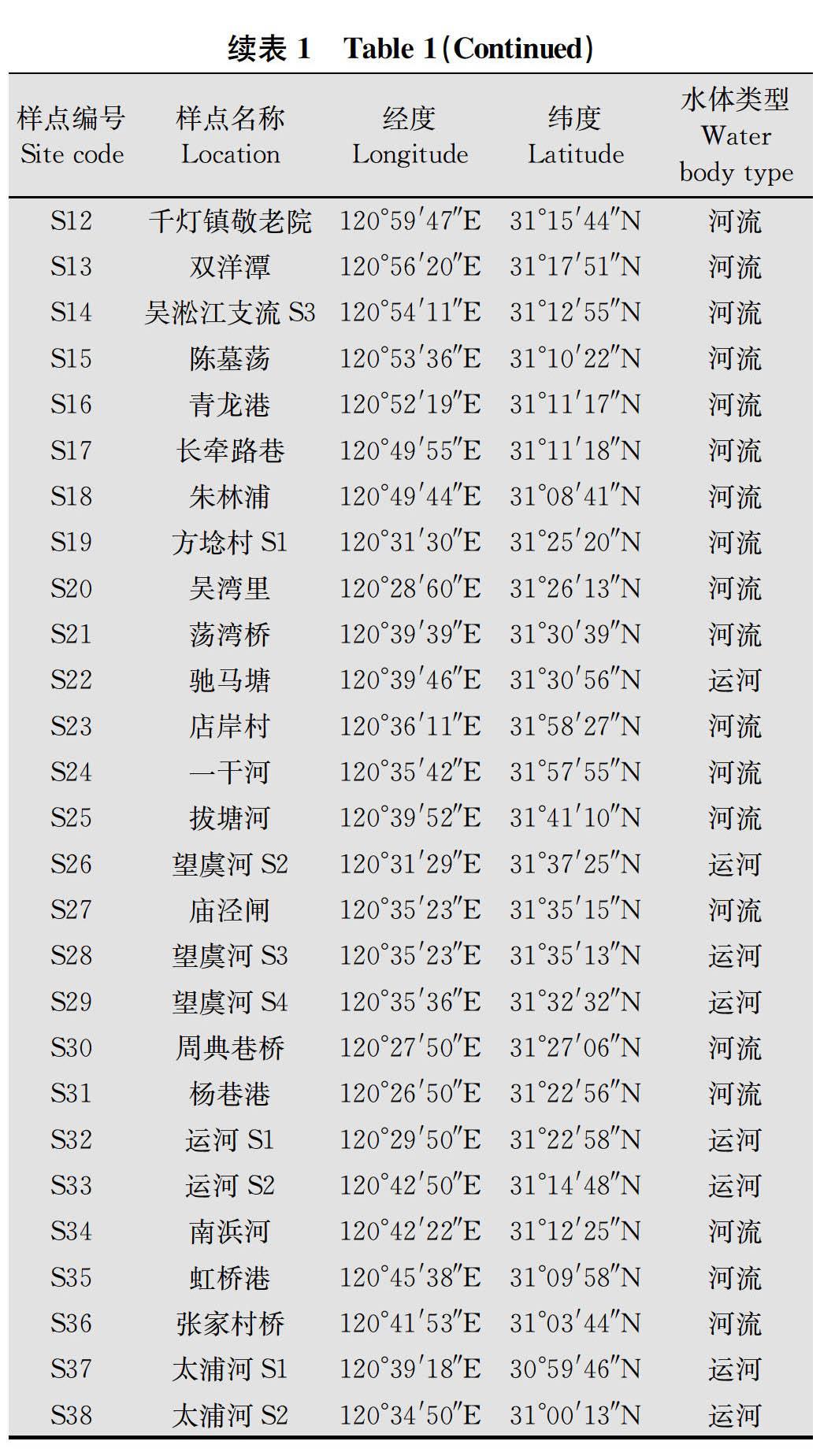
苏州地区处于江苏省南部,属于长江流域太湖水系,水系发达,河网稠密,为福寿螺的繁殖和扩散提供了良好条件[18]。本研究于2019年1月在苏州市相城区、常熟市、张家港市及昆山市进行水环境样品及福寿螺生物样品的采集,共设置了38个样点(编号S01~S38),其中河流样点24个,湖泊样点6个和运河样点8个(表1)。
1.2 环境DNA采集和福寿螺发生调查
采用过滤法收集水体中的环境DNA。每个样点设3个采样位置,各位置相隔5 m,每个位置在30~50 cm水深处用采样瓶各采集500 mL水样,合计1.5 L。将玻璃微纤维滤膜(GF/F,孔径0.7 μm)放置于过滤装置(Nalgene 300-4100过滤器)中,随后将充分摇匀的水样通过过滤装置进行过滤。水样过滤后用镊子将滤膜放入离心管中,并加入无水乙醇置于4℃移动冰箱短期保存,带回实验室后放在-20℃环境保存。每次过滤前用75%乙醇和超纯水充分冲洗过滤装置,以防止各样点交叉污染,所有仪器和材料在使用前均灭菌处理。
采用现场观察法记录采样点周边100 m范围内福寿螺的发生情况,观察到有空壳、卵块、活体的水域均认为有福寿螺存在,并随机采集10只福寿螺活体带回实验室。
1.3 DNA提取与扩增
将38个样点分3组进行环境DNA提取与扩增,每组包括12(或13个)样点收集的样品以及1个阳性对照和1个阴性对照。阳性对照(SP)为10头冲洗干净的福寿螺在1.5 L无菌水内培养72 h后用过滤法收集的环境DNA样本,使用纯净水过滤后的样本作为阴性对照(SN)。另外选取4头活体福寿螺,分别提取其组织DNA后测序获得物种COⅠ序列,构建本地福寿螺序列数据库。试验结束后10头福寿螺均用75%乙醇杀死并作为标本保存。
环境DNA扩增前首先用无菌剪刀将滤膜剪碎成约3 mm的碎片,使用上海生工Ezup柱式动物基因组DNA抽提试剂盒(Ezup Column Animal Genomic DNA Purification Kit)从滤膜中提取环境DNA。采用上游引物mlCOlintF:5′-GGWACWGGWTGAACWGTWTAYCCYCC-3′和下游引物jgHCO2198:5′-TANACYTCNGGRTGNCCRAARAAYCA-3′(用N替代原序列中的I)对样品的环境DNA进行扩增[19]。在每对引物上游、下游的5′-末端分别添加8个核苷酸长度的标签,用以区分各个测序组中的每个样品。PCR反应体系为50 μL。反应条件为94℃ 5 min;95℃ 10 s,62℃ 30 s(每次循环该过程都比上1个循环降低1℃),72℃ 60 s,16个循环;95℃ 10 s,46℃ 30 s,72℃ 30 s,25个循环;72℃ 5 min。测序平台为Illumina Hiseq 2 500。
福寿螺组织DNA提取试剂盒同上,利用上游引物LCO1490:5′-GGTCAACAAATCATAAAGATATTGG-3′和下游引物HCO2198: 5′-TAAACTTCAGGGTGACCAAAAAATCA-3′进行扩增[20]。反应条件为94℃ 1 min;94℃ 30 s,45℃ 90 s,72℃ 1 min,6个循环;94℃ 30 s,51℃ 90 s,72℃ 1 min,36个循环;72℃ 5 min。测序平台为ABI 3730XL。
1.4 数据处理与分析
利用SEED软件(Version 2.1.05)对测序获得的原始数据进行处理与分析,将原始数据成对导入并进行拼接组装[21]。筛选出质量Q≥30的序列,在去除过长和过短序列后,按照标签将序列分组为各个样点数据。去除引物与标签不匹配的错误序列,删去嵌合体。对所得优质序列按照大于97%的相似度进行可操作分类单元(operational taxonomic units, OTUs)聚类。将获得的OTUs序列与NCBI上的数据库进行BLAST比对。为防止NCBI中发布的序列错误或比对出现错误,我们同时将OTUs与基于福寿螺组织DNA COⅠ序列构建的本地序列数据库进行比对,完成OTUs的物种信息注释。将经过处理分析后的信息汇总,得到各样点中监测到的福寿螺OTUs的丰度表,筛选其中一致性(similarity)=100%,覆盖度(coverage)=100%的结果为有效的可操作分类单元。
为了评估低丰度序列的准确性,我们设置了序列丰度阈值和相对丰度阈值两种筛选方式,研究不同阈值设置对检测结果的影响[22]。序列丰度(n)即每个OTU中各个样点测得的序列数。相对丰度(m)是指某样点的目标OTU序列数占该样点检测到的所有OTUs序列总数的比例。将得到的物种序列数进行标准化处理,通过lg(n+1)进行对数转换。所有的分析及作图通过R语言(Version 4.0.1)完成。利用R语言中的“pheatmap包”对处理后的小管福寿螺序列及每个样点的总序列数据进行对比统计,制作丰度热图。为评估相对序列丰度是否合适作为筛选物种存在的方式,将标准化处理的数据利用R语言中的“lm包”进行线性拟合。为探讨两种方法检测到的福寿螺发生率是否与水体类型有关,我们进一步分析了河流、湖泊、运河中的福寿螺检出率,即环境DNA技术检测到福寿螺序列或现场观测到福寿螺的样点数与检测总样点数的比值。
2 结果与分析
2.1 现场观察法和环境DNA-宏条形码技术检测结果
现场观察法在38个样点中的14个样点观察到小管福寿螺发生痕迹,其中活体、卵块和空壳都观察到的有2个样点(S19、S23),观察到卵块和空壳的有4个样点(S20、S21、S35、S36),仅观察到卵块或空壳的样点数分别有6个(S08、S16、S31、S34、S37、S38)和2个(S12、S15)。
利用环境DNA-宏条形码技术在38个样点和6组对照中共得到21 973 306条DNA序列,其中福寿螺的序列数共111 317條。在36个样点中检测到4 852条福寿螺序列,阳性对照组(SP)检测到106 465条福寿螺序列,阴性对照组(SN)未检测到福寿螺序列,说明试验操作无污染。通过USEARCH聚类得到两个OTU,经比对及注释,其GenBank序列号分别为EF514972和EU528586,均为小管福寿螺。其中36个样点及3个阳性对照中共检测到103 141条序列的注释结果为EF514972,远高于注释结果为EU528586的8 176条序列(表2,图1)。此外,从现场采集的福寿螺中获得的COⅠ序列经BLAST比对及注释后结果同样为小管福寿螺,验证了环境DNA技术检测到的物种信息结果准确。
2.2 不同序列丰度阈值对检测结果的影响
不同序列丰度阈值下小管福寿螺COⅠ条码序列检出率的结果(表3)表明,在序列丰度阈值n≥3和相对丰度阈值m≥0.001%两种阈值下检测到小管福寿螺序列的样点数一致。其中,环境DNA技术检测到小管福寿螺COⅠ序列的样点数为35个,占总调查样点的92.11%,在14个现场调查到福寿螺个体的样点中有13个检测到小管福寿螺COⅠ序列。但随着序列丰度阈值和相对丰度阈值的提高,检出率随之下降。当序列丰度阈值n≥15时,小管福寿螺COⅠ序列的检出率仅为57.89%,14个调查到福寿螺个体样点中仅有7个样点(50%)检测到小管福寿螺序列;而序列相对丰度阈值m≥0.030%时,小管福寿螺COⅠ序列的检出率仅为47.37%,14个调查到福寿螺个体样点中仅有5个样点(35.71%)检测到小管福寿螺COⅠ序列。
基于38个样点及对照组共44个样本的总序列数及小管福寿螺序列数的线性拟合结果显示,总序列数与小管福寿螺序列数间不存在线性关系(P>0.05,图2),这表明基于序列相对丰度阈值m检测到的小管福寿螺发生率不能够真实地反映福寿螺的实际发生情况。结合表3的结果,本文在后续分析中,以序列丰度阈值n作为环境DNA技术检测小管福寿螺发生的指标。
2.3 不同水体类型中小管福寿螺的检测结果
当序列丰度阈值n≥2与n≥3时,从环境DNA中检测到小管福寿螺的结果与现场观察的结果一致(表3),且此时两种方法检测到小管福寿螺的样点数均最多,现以序列丰度阈值n≥3作为小管福寿螺COⅠ条形码检出的阈值来计算其在不同水体类型中的检出率(表4)。结果表明,湖泊中小管福寿螺检出率最高,达到100%,河流和运河检出率分别为91.67%和87.50%。但是,现场观察法并未在6个湖泊样点中调查到小管福寿螺,而24个河流样点中有12个样点(50%)调查到小管福寿螺,8个运河样点中有2个样点(25%)调查到小管福寿螺。综上所述,38个调查样点中,现场观察法成功调查到小管福寿螺的共14个样点(36.84%),远低于从环境DNA中检测出小管福寿螺的35个样点(92.11%)。
3 讨论
目前我国报道有3种外来入侵福寿螺,即小管福寿螺、斑点福寿螺Pomacea maculata和Pomacea sp.,其中小管福寿螺与斑点福寿螺亲缘关系近,形态与生活习性也相近,利用传统分类方法很难区分二者[4]。基于DNA条形码技术研究发现,云南省、浙江省均已监测到斑点福寿螺的入侵[4,23]。然而,此前的研究中,苏州地区仅发现小管福寿螺,尚不确定是否有其他福寿螺入侵物种存在[24]。本文通过分子技术验证了苏州地区现阶段仅有小管福寿螺一种入侵物种存在。
针对入侵物种小管福寿螺的检测,环境DNA-宏条形码技术相比传统观察法更加灵敏。在38个样点中环境DNA技术在序列丰度阈值n≥1时检测到小管福寿螺的样点数有36个,明显高于现场观察法(14个)。当序列丰度阈值设置为n≥3,环境DNA技术检测到小管福寿螺的发生率(92.11%)远高于传统观察法(36.84%)。然而,利用环境DNA技术检测也存在假阴性结果。现场观察到小管福寿螺的14个样点中S21样点在现场观察到福寿螺卵块及空壳,但环境DNA技术并未检测出,这可能与其在环境中的DNA含量少有关,处于不同发育阶段的生物其环境DNA释放量存在差异[25],也有可能是生物信息分析过程中由于筛选标准高,序列被删除而造成的假阴性结果[26]。
环境DNA技术中的生物信息学数据处理是影响结果的重要步骤。低丰度序列可能是人为因素或误差产生的错误序列,尤其是单体序列(singleton, n=1)[27]。设置丰度阈值是降低低丰度序列造成假阳性结果的一种常用方法[28]。但也不可否认在检测低丰度物种时,低丰度序列可能反映了物种存在的重要信息[29]。在已有研究中,一般存在两种筛选方法,一种是根据序列丰度阈值筛选[30],一种是根据序列相对丰度阈值[31]来去除低丰度序列的OTUs。本文研究了上述两种丰度阈值对小管福寿螺检测结果的影响,结果表明,从单个环境DNA样本中检测出小管福寿螺的序列数与总序列数没有显著相关性,而序列相对丰度阈值是基于总序列数计算得到的,说明基于序列相对丰度阈值m的结果并不能准确反映小管福寿螺的入侵,针对低丰度生物的检测时采取序列丰度阈值可能更合适。小管福寿螺序列数为n=1的样点只有S22,在该样点我们并未观察到其物种存在痕迹,所以我们认为该结果为假阳性结果,即与实际不相符。本研究中,序列丰度阈值为n≥3时利用环境DNA技术在35个样点中检测到小管福寿螺COⅠ条形码。关于n值多少为最佳,针对不同物种阈值设置是否不同,仍需要进一步研究。
环境DNA技术和现场观察法在不同水体类型中的检测结果也存在差异。在所有的湖泊样点中,环境DNA技术都检测到了小管福寿螺,这与我们前期在这些湖泊中发现有小管福寿螺分布的事实相符,而传统观察法在湖泊水体并未观测到小管福寿螺。湖泊水体在监测时受水深、生物习性等因素影响,而传统观察及捕捞多在沿岸区域进行,底栖性的小管福寿螺在监测时,极有可能难以发现。在运河水体中,利用环境DNA技术的检出率(87.50%)也高于传统观察法(25%)。相比传统观察法只在河流水体中有较高的检出率不同,环境DNA技术在3种水体类型中都有较高的检出率,表明该方法受水体类型的影响较小。
本文的研究结果表明环境DNA技术对入侵水生生物的监测有重要的应用价值,该技术可以高效敏捷地监测小管福寿螺的入侵分布。虽然该技术仍存在低丰度物种检出率低,且不能定量的不足[32],但环境DNA技术具有可对大量的混合样本快速检测,受时间和环境因素影响小,对生物和环境近乎无伤,操作简单便捷,省时省力灵敏度高等优点。相比传统观察法,环境DNA-宏条形码技术可在福寿螺入侵初期进行监测,且受水体类型影响小。相信随着环境DNA的获取及数据分析方法的标准化及优化,其必将成为未来入侵水生生物检测的优选方法。
参考文献
[1] RICHARD N M, DANIEL S W, MARK L, et al. Biotic invasions: causes, epidemiology, global consequences, and control [J]. Ecological Applications, 2000, 10(3): 689710.
[2] LOWE S, BROWNE M, BOUDJELAS S, et al. 100 of the world’s worst invasive alien species: a selection from the global invasive species database[C]∥Invasive Species Specialist Group of the World Conservation Union. Auckland, New Zealand: ISSG Publications, 2000.
[3] 李小慧, 胡隐昌, 宋红梅, 等. 中国福寿螺的入侵现状及防治方法研究进展[J]. 中国农学通报, 2009, 25(14): 229232.
[4] 杨倩倩, 刘苏汶, 茹炜岽, 等. 基于DNA条形码技术对浙江省外来入侵福寿螺进行分子鉴定[J]. 生物多样性, 2016, 24(3), 341350.
[5] 周宇, 袁雪颖, 杨子轩, 等. 福寿螺入侵中国的扩散动态及潜在分布[J]. 湖泊科学, 2018, 30(5): 13791387.
[6] 中国环境保护总局. 中国第一批外来入侵物种名单[EB/OL]. (20030110) [20200704]. http:∥www.mee.gov.cn/gkml/zj/wj/200910/t20091022_172155.htm.
[7] 查国贤, 李俊, 沈晴, 等. 苏州市农业植物检疫性有害生物发生现状及防控对策[J]. 植物检疫, 2018, 32(5): 7579.
[8] TABERLET P, COISSAC E, HAJIBABAEI M, et al. Environmental DNA [J]. Molecular Ecology, 2012, 21(8): 17891793.
[9] 陈炼, 吴琳, 刘燕, 等. 环境DNA metabarcoding及其在生态学研究中的应用[J]. 生态学报, 2016, 36(15): 45734582.
[10]JERDE C L, MAHON A R, CHADDERTON W L, et al. ‘Sight-unseen’ detection of rare aquatic species using environmental DNA[J]. Conservation Letters, 2011, 4(2): 150157.
[11]JERDE C L, CHADDERTO W L, MAHON A R, et al. Detection of Asian carp DNA as part of a Great Lakes basin-wide surveillance program [J]. Canadian Journal of Fisheries and Aquatic Sciences, 2013, 70(4): 522526.
[12]TAKAHARA T, MINAMOTO T, YAMANAKA H, et al. Estimation of fish biomass using environmental DNA [J/OL]. PLoS ONE, 2012, 7(4): e35868. DOI: 10.1371/journal. pone.0035868.
[13]馬竹欣. 利用环境DNA技术调查入侵种克氏原螯虾在元阳梯田的分布[D]. 昆明: 云南大学, 2016.
[14]刘雨芳, 李菲, 李玉峰, 等. 福寿螺在湖南的分布现状、危害与扩散风险预警[J]. 水生生物学报, 2011, 35(6): 10671071.
[15]周兵, 谢春扬迪, 闫小红, 等. 江西省福寿螺的入侵危害现状及稻田发生规律[J]. 生态与农村环境学报, 2015, 31(6): 902909.
[16]刘义满, 戢小梅, 李长林, 等. 湖北省福寿螺分布调查[J]. 长江蔬菜, 2020(6): 5765.
[17]FEDIAJEVAITE J, PRIESTLEY V, ARNOLD R, et al. Meta-analysis shows that environmental DNA outperforms traditional surveys, but warrants better reporting standards [J]. Ecology and Evolution, 2021, 11(9): 48034815.
[18]蘇州市统计局. 苏州统计年鉴—2019[M]. 北京: 中国统计出版社, 2019: 45.
[19]LERAY M, YANG J Y, MEYER C P, et al. A new versatile primer set targeting a short fragment of the mitochondrial COⅠ region for metabarcoding metazoan diversity: application for characterizing coral reef fish gut contents [J]. Frontiers in Zoology, 2013, 10(1): 34. DOI: 10.1186/1742-9994-10-34.
[20]FOLMER O, BLACK M, HOEH W, et al. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates [J]. Molecular Marine Biology and Biotechnology, 1994, 3(5): 294299.
[21]VETROVSKY T, BALDRIAN P, MORAIS D. SEED 2: a user-friendly platform for amplicon high-throughput sequencing data analyses [J]. Bioinformatics, 2018, 34(13): 22922294.
[22]ZHAN Aibin, XIONG Wei, HE Song, et al. Influence of artifact removal on rare species recovery in natural complex communities using high-throughput sequencing [J/OL]. PLoS ONE, 2014, 9(5): e96928. DOI: 10.1371/journal.pone.0096928.
[23]冉甄. 基于线粒体COⅠ基因对云南省不同水系外来入侵生物福寿螺的系统发育学研究[D].昆明: 昆明医科大学, 2019.
[24]YANG Qianqian, LIU Suwen, HE Chao, et al. Invisible apple snail invasions: importance of continued vigilance and rigorous taxonomic assessments [J]. Pest Management Science, 2019, 75(5): 12771286.
[25]CHRISTOPHER R T, MICHAEL J S, JEN N, et al. Seasonality, DNA degradation and spatial heterogeneity as drivers of eDNA detection dynamics [J/OL]. Science of The Total Environment, 2021, 768: 144466. DOI: 10.1016/j.scitotenv.2020.144466.
[26]FURLAN E M, DAVIS J, DUNCAN R P. Identifying error and accurately interpreting eDNA metabarcoding results: A case study to detect vertebrates at arid zone waterholes [J]. Molecular Ecology Resources, 2020, 20(5): 12591276.
[27]KUNIN V, ENGELBREKTSON A, OCHMAN H, et al. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates [J]. Environmental Microbiology, 2010, 12(1): 118123.
[28]GIGUET-COVEX C, PANSU J, ARNAUD F, et al. Long livestock farming history and human landscape shaping revealed by lake sediment DNA [J/OL]. Nature Communications, 2014, 5: 4211. DOI: 10.1038/ncomms4211.
[29]ZHAN Aibin, HULK M, SYLVESTER F, et al. High sensitivity of 454 pyrosequencing for detection of rare species in aquatic communities [J]. Methods in Ecology and Evolution, 2013, 4(6): 558565.
[30]CAREW M E, PETTIGROVE V J, METZELING L, et al. Environmental monitoring using next generation sequencing: rapid identification of macroinvertebrate bioindicator species [J/OL]. Frontiers in Zoology, 2013, 10(1): 45. DOI: 10.1186/1742-9994-10-45.
[31]ELBRECHT V, LEESE F. Can DNA-based ecosystem assessments quantify species abundance? Testing primer bias and biomass-sequence relationships with an innovative metabarcoding protocol [J/OL]. PLoS ONE, 2015, 10(7): e0130324. DOI: 10.1371/journal.pone.0130324.
[32]李杰, 杨婧, 黄原. 应用DNA复合条形码技术研究秦岭水生动物多样性[J]. 生态学报, 2016, 36(19): 61036112.
(责任编辑:杨明丽)
