高效液相色谱-串联质谱法快速测定兽药制剂中喹诺酮类、磺胺类等26种药物
2021-01-01刘秀娟冯慧慧杨亚琴余明霞董小海钟红舰
曹 秀,刘秀娟,冯慧慧,杨亚琴,余明霞,董小海,钟红舰
(河南省农业科学院农业质量标准与检测技术研究所,郑州 450002)
目前,国内兽药市场竞争激烈,兽药制剂中非法添加化学药物的现象时有发生,添加药物种类多样,如禁用药物、人用药物;添加手段隐蔽,如同类药物代替标称药物,使得兽药检测与监管的难度越来越大[1-2]。为进一步严格兽药生产与管理,农业农村部近年来连续发布了关于《兽药中非特定非法添加物质检查方法》的公告作为兽药非法添加的法定标准[3-4]。兽药非特定非法添加物质的检查方法主要有显微镜检查法[5]、薄层色谱法[6]、高效液相色谱法[7-10]、液相色谱串联质谱法[11-14]等。显微镜检查法适用性范围窄;薄层色谱法是初筛的常见手段,但存在假阳性的风险。高效液相色谱配二极管阵列检测法(HPLC-PDA)应用范围广泛,适合于不同种类兽药制剂的非法药物添加检测,能准确定性和定量。该方法日益成熟,农业农村部颁布诸多公告多为该方法。然而,HPLC-PDA在兽药非法添加物质检查中存在局限性,如无法鉴别PDA光谱图库以外的化合物;部分化合物在不同色谱条件下光谱图差异大,难溯源;无法检测无紫外吸收的化合物,造成漏筛等[15]。液相色谱-串联质谱法由于其高特异性、高灵敏度的优点,已成为检测违禁添加药物的重要手段。该方法对添加物的检测无歧视,在合适的质谱条件下,可以获得其分子离子及碎片的信息,对推断未知物很有尤为重要,LC-MS/MS通过定性离子与定量离子能对未知物进行准确定量。近年来,LC-MS/MS技术在兽药制剂中氯霉素、硝基呋喃类药物、磺胺类药物、硝基咪唑类药物及喹诺酮类药物的检测均有报道[11-14]。
喹诺酮类及磺胺类药物是水产和畜牧养殖中常用的广谱抗菌药物,也是常见的非法添加药物;孔雀石绿、金刚烷胺与金刚乙胺属于禁用药物,这些药物在动物体内残留及代谢,会对人类健康造成一定伤害。兽药制剂基质种类多,如预混剂、可溶性粉剂、兽药散剂、注射液等,不能直接采用常规的饲料或动物源中兽药残留检测分析技术进行检测,因此建立一套有效的兽药制剂中多种兽药残留分析方法非常重要。
研究建立了兽药制剂中喹诺酮类、磺胺类、孔雀石绿、金刚烷胺等26种药物的高效液相色谱串联质谱法,该方法简单快捷,灵敏度高、结果准确可靠,可满足市场兽药制剂中多种药物的筛查需求,为兽药监管提供有效的技术支持。
1 材料与方法
1.1 仪器 LC MS-8050 高效液相色谱-串联质谱仪,配有电喷雾离子源及LabSolution LC MS数据处理系统(日本Shimadzu公司);快速混匀器SK-1(金坛市中大仪器厂);高速冷冻离心机GL-21B(上海安亭科学仪器厂);氮吹仪QGC-36T(上海全岛公司)。
1.2 药品与试剂 氧氟沙星、诺氟沙星、甲磺酸培氟沙星、盐酸环丙沙星、洛美沙星、甲磺酸达氟沙星、恩诺沙星、盐酸沙拉沙星、氟甲喹、磺胺嘧啶、磺胺噻唑、磺胺甲基嘧啶、磺胺甲噻二唑、磺胺二甲嘧啶、磺胺氯哒嗪、磺胺间甲氧嘧啶、磺胺甲基异噁唑、磺胺二甲异噁唑、磺胺邻二甲氧嘧啶、磺胺间二甲氧嘧啶、磺胺喹恶啉、甲氧苄啶、孔雀石绿、隐色孔雀石绿、金刚烷胺、金刚乙胺,26种化合物标准物质均购自德国Dr.Ehrenstorfer 公司,纯度均大于99.0%。甲醇、乙腈、甲酸,色谱纯,购自德国Mecrk公司。实验所用水为经Milli-Q净化系统(美国Millipore公司)制备的超纯水。试验所用兽药制剂样品均为市售合格样品,分别为20%氟苯尼考粉、30%氟苯尼考可溶性粉、10%恩诺沙星可溶性粉、25%维生素C可溶性粉。
1.3 色谱及质谱条件
1.3.1 液相色谱条件 色谱柱CAPCELL PAK C18(100 mm×2.0 mm,3 μm);柱温40 ℃;流动相A相为5 mmol/L甲酸-甲酸铵,B相为甲醇-乙腈(1∶1,V∶V);线性梯度洗脱程序:0~2 min,90% A;2~3 min,85% A;3~13 min,85% A;13~20 min,15%A;20~25 min,15% A;25.10 min恢复初始比例,90% A,保持至28 min;流速为0.200 mL/min;进样体积为1 μL。
1.3.2 质谱测定条件 电喷雾离子源,正离子扫描;多反应监测模式;离子源接口电压4.5 kV;雾化气流速3 L/min;干燥气流速10 L/min;加热器流速10 L/min;脱溶剂管温度250 ℃;加热模块温度400 ℃;接口温度300 ℃;碰撞诱导电离气体压力270 kPa,各化合物质谱采集参数见表1。
1.4 标准溶液的制备
1.4.1 单标标准储备液的制备 分别精密称取5 mg(精确至0.01 mg)26种化合物标准物质于各自的5 mL容量瓶中,用甲醇溶解并定容至刻度,配制成1 mg/mL的单标标准储备溶液,于-20 ℃以下避光冷冻保存。
1.4.2 26种化合物混合标准储备液的配制 分别精密称取5 mg(精确至0.01 mg)26种化合物标准物质于5 mL容量瓶中,用甲醇溶解并定容至刻度,配制成1 mg/mL的26种化合物混合标准储备溶液,于-20 ℃以下避光冷冻保存。
1.4.3 26种化合物混合标准工作液的配制 准确移取0.1 mL 26种化合物混合标准储备液(1 mg/mL)至10 mL容量瓶中,用甲醇溶解并定容至刻度,配制成 10 μg/mL的26种化合物混合标准工作液,于4 ℃避光冷藏保存。
1.4.4 26种化合物混合标准曲线的配制 精密移取适量26种化合物10 μg/mL混合标准工作液,用甲醇水(1∶1,V∶V)溶液稀释,配制成质量浓度分别为0.1、0.2、1、2、10 ng/mL的混合标准工作曲线。
1.5 兽药制剂前处理 准确称取1.00 g(精确至0.01 g)兽药制剂于50 mL塑料离心管中,加10 mL 0.2%甲酸乙腈超声提取10 min,置于振荡器振荡30 min,8000 r/min离心5 min,移取2 mL上清液于氮气吹干仪中浓缩至干,用1 mL甲醇溶解残渣,加1 mL 0.2% 甲酸水溶液,涡旋混匀,过0.22 μm微孔滤膜得到预备液。为避免兽药原药对质谱的污染,预备液经甲醇:水溶液(1∶1,V∶V)稀释10000倍后,经LC-MS/MS测定。
1.6 检出限与定量限 选取20%氟苯尼考粉、30%氟苯尼考可溶性粉作为空白样品,在空白样品添加适量的26种化合物混合标准储备液,按1.5方法处理样品,经1.3方法仪器条件进行测定。依据特征离子质量色谱峰信噪比S /N≥3 的浓度为方法检出限(LOD),S /N≥10 的浓度为方法定量限(LOQ)。
1.7 精密度与准确度 采用标准添加法进行加标回收实验。分别在20%氟苯尼考粉、30%氟苯尼考可溶性粉样品中添加LOD、LOQ及5倍LOQ三个浓度水平的26种化合物混合标准储备液,添加浓度见表3。按1.5方法处理样品,经1.3方法仪器条件进行测定。每个添加浓度进行5个样品的平行试验,计算平均回收率及相对标准偏差。
1.8 实际样品的测定方法 选取市售合格的兽药制剂样品,分别为20%氟苯尼考粉、30%氟苯尼考可溶性粉、10%恩诺沙星可溶性粉、25%维生素C可溶性粉按照1.5方法处理,经1.3方法仪器条件进行测定。
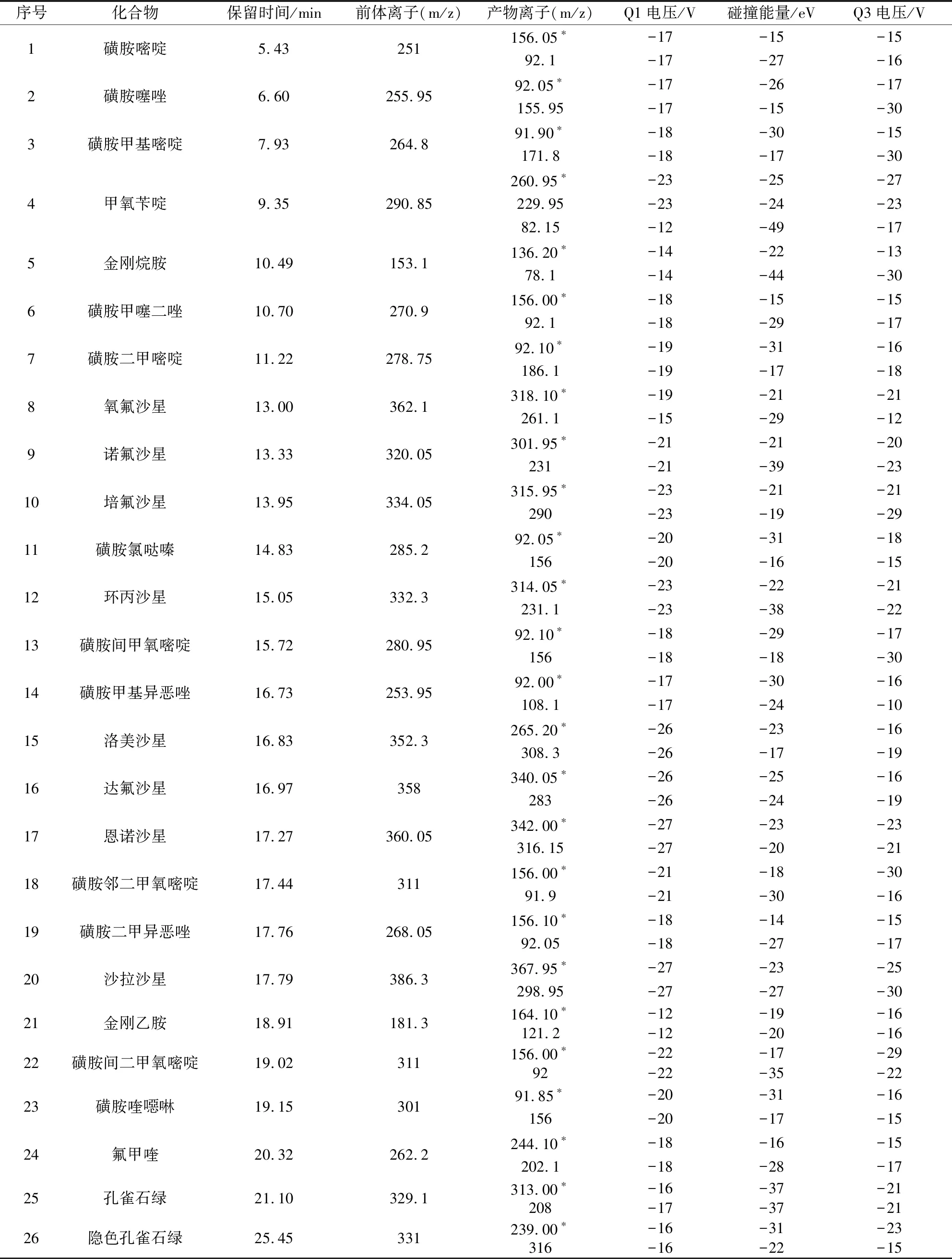
表1 26种化合物质谱参数及保留时间Tab 1 Retention time and main mass spectrometry parameters of 26 compounds
2 结果与分析
2.1 线性范围 按1.4.4方法配制的26种化合物混合标准工作曲线在1.3方法仪器条件下测定。26种化合物混合标准溶液(10 ng/mL)总离子流图见图1,26种化合物出峰顺序及保留时间见表1,26种化合物分离度良好,响应灵敏。以各化合物定量离子色谱峰面积A为纵坐标,各化合物质量浓度(ng/mL)为横坐标,绘制26种化合物的线性回归方程。结果表明:26种化合物在0.1~10 ng/mL浓度范围内线性关系良好,相关系数r2均大于0.99,26种化合物的线性回归方程及相关系数见表2。
2.2 检出限与定量限 20%氟苯尼考粉、30%氟苯尼考可溶性粉作为空白样品经1.5方法处理后,测定结果表明:在相应的保留时间,空白样品及试剂空白对26种目标化合物无干扰,空白样品图谱及试剂空白图谱见图2。依据特征离子质量色谱峰信噪比S /N≥3 的浓度为方法检出限,该方法检出限为0.01 mg/g,特征离子质量色谱峰信噪比S /N≥10 的浓度为方法定量限,方法定量限为0.02 mg/g。
2.3 精密度与准确度 依据1.7方法制备添加浓度分别为0.01 mg/g、0.02 mg/g、0.1 mg/g的阳性样品,每个浓度水平下平行测定5次,外标法标准曲线定量,26种化合物的平均回收率为70.0%~101%,相对标准偏差(RSD)为1.9%~8.8%,结果见表3。图3为添加浓度为0.1 mg/g兽药制剂阳性样品图谱。
2.4 实际样品的测定 对市售的20%氟苯尼考粉、30%氟苯尼考可溶性粉、10%恩诺沙星可溶性粉、25%维生素C可溶性粉分别进行HPLC-MS/MS检测。结果表明:20%氟苯尼考粉、30%氟苯尼考可溶性粉、25%维生素C可溶性粉均未检出26种化合物。10%恩诺沙星可溶性粉兽药制剂除标称“恩诺沙星”检出外,其余25种化合物未检出。经测定,10%恩诺沙星可溶性粉兽药制剂中恩诺沙星含量为105 mg/g(有效含量为10.5%),与产品标识含量(10%恩诺沙星)一致。图4为10%恩诺沙星可溶性粉图谱。
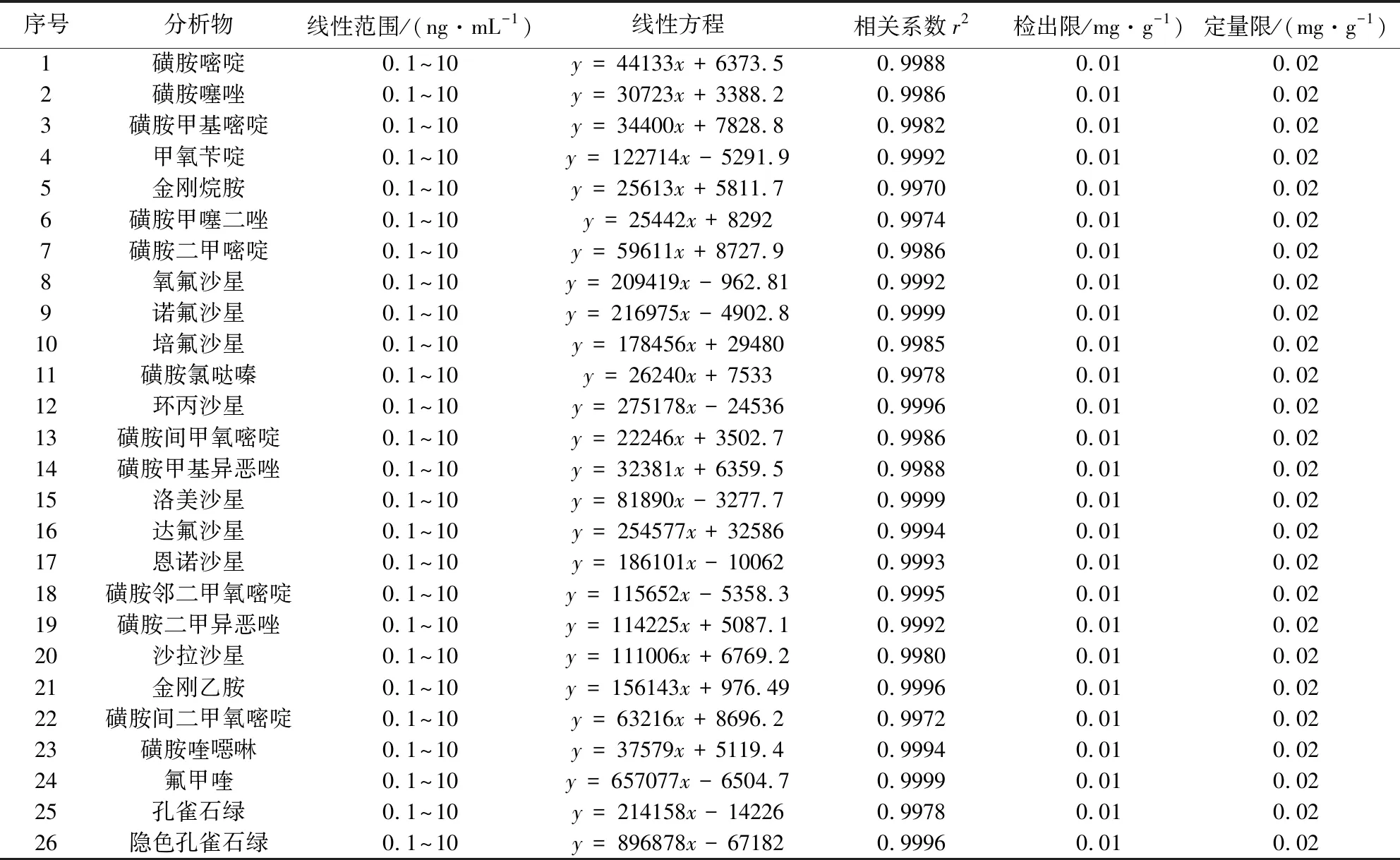
表2 26种化合物线性范围、线性方程、相关系数、检出限及定量限Tab 2 Regression equation,correlation coefficients(r2),LODs and LOQs of 26 compounds
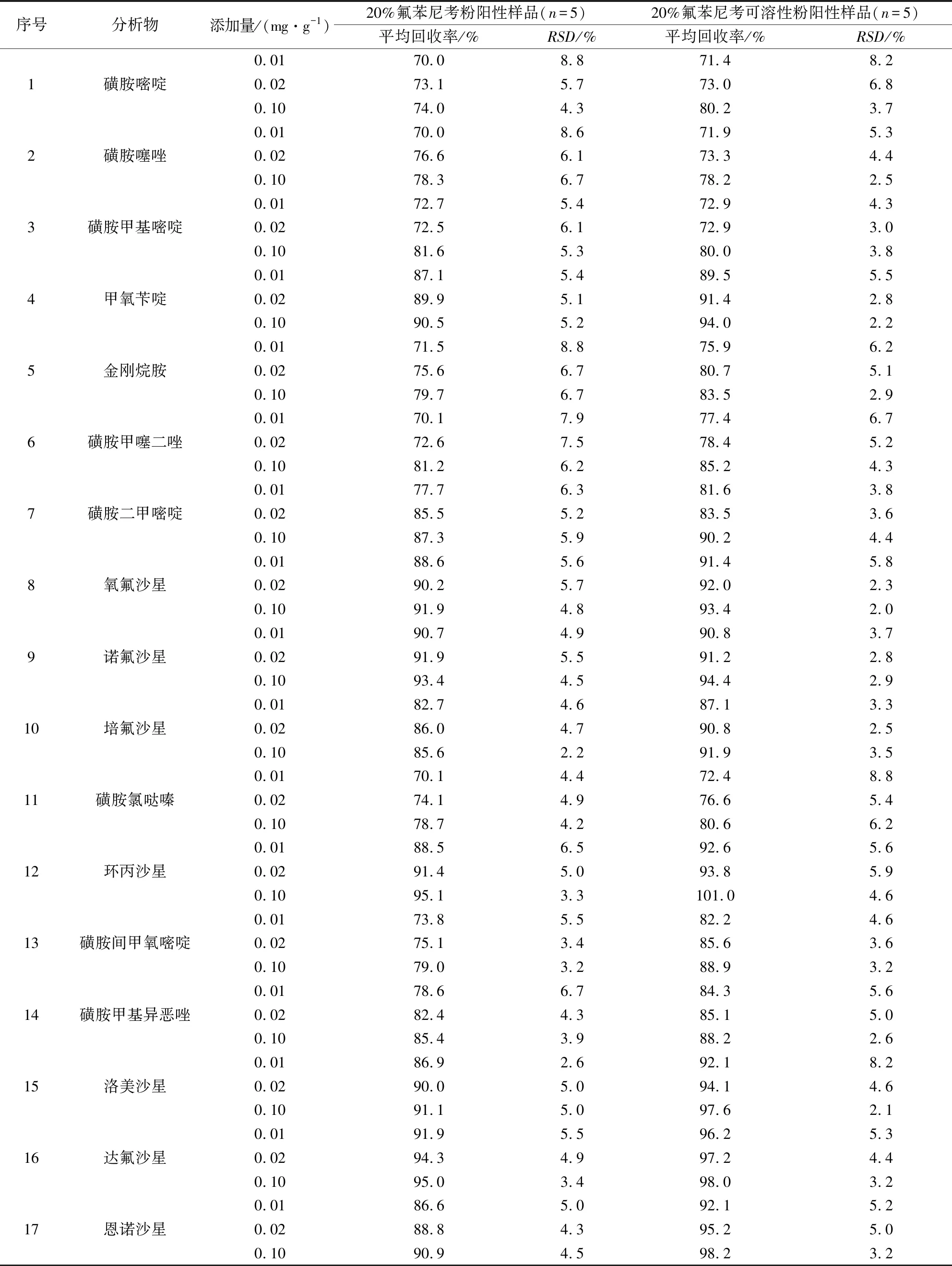
表3 26种化合物在阳性样品中的回收率(n=5)Table3 Recoveries of 26 compounds in different positive samples (n=5)

续表3

图1 26种化合物混合标准溶液(10 ng/mL)总离子流图Fig 1 Total ion flow chromatograms of 26 compounds (10 ng/mL)

图2 (A)20%氟苯尼考粉样品空白(B)30%氟苯尼考可溶性粉样品空白(C)溶剂空白图谱Fig 2 Chromatograms of blank samples (A) 20% florfenicol powder (B) 30% florfenicol soluble powder)and(C) solvent blank
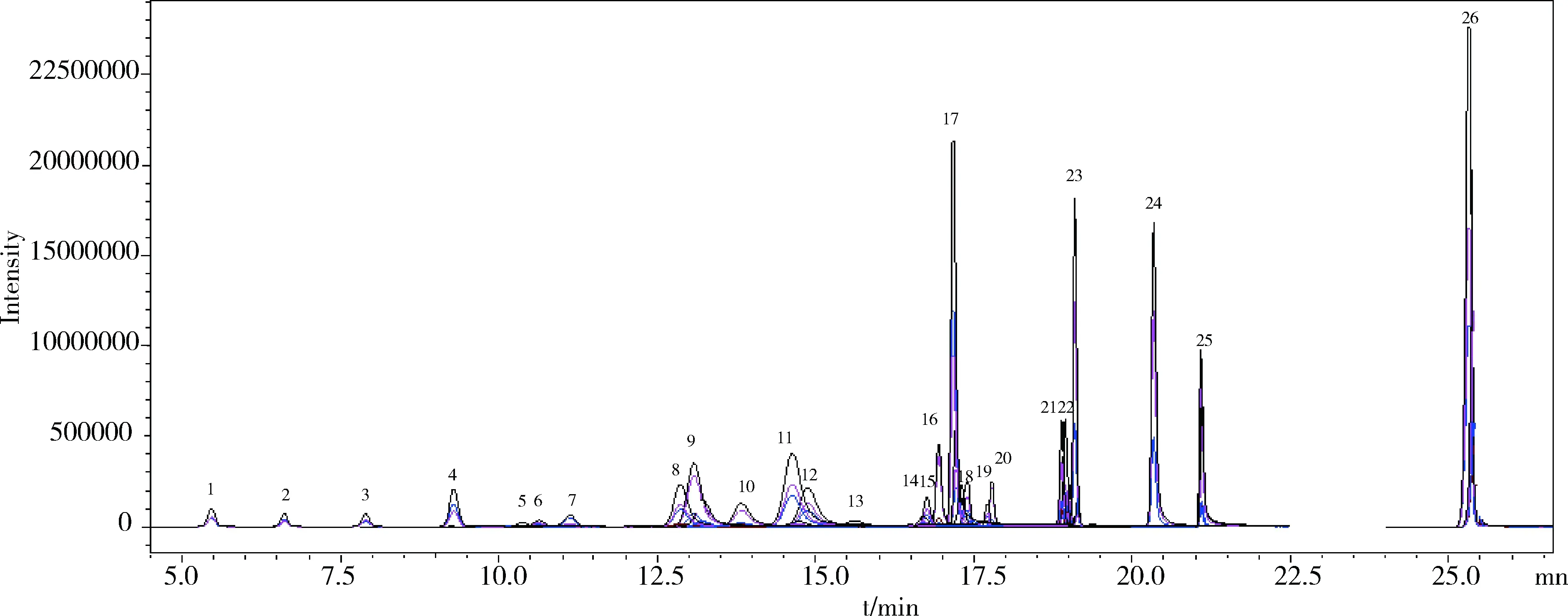
图3 兽药制剂阳性样品(添加量为0.1 mg/g)图谱Fig 3 Chromatograms of spiked sample(0.1 mg/g)
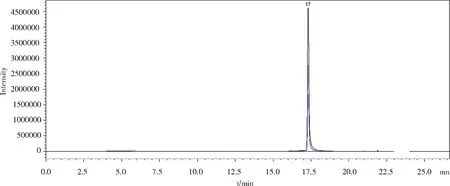
图4 实际样品检测(10%恩诺沙星可溶性粉)图谱Fig 4 Actual sample test (10% enrofloxacin soluble powder)
3 讨论与结论
3.1 色谱条件的优化 研究选用CAPCELL PAK C18(100 mm×2.0 mm,3 μm)色谱柱对26种化合物进行分离,依据目标化合物的性质,流动相均选用酸性溶液,如0.1%甲酸水,乙酸铵溶液、甲酸-甲酸铵复配溶液。本研究考察了甲醇-乙腈(1∶1,V∶V)作为有机相时,选取0.1%甲酸水、5 mmol/L乙酸铵及5 mmol/L甲酸-甲酸铵作为水相,采用梯度洗脱的方式对26种化合物进行分离。研究结果表明,当选用5 mmol/L甲酸-甲酸铵作为水相时,26种化合物的分离度及峰形优于0.1%甲酸水和5 mmol/L 乙酸铵溶液。
3.2 质谱条件的优化 方法采用不接分析柱的方式向质谱系统注入 1 μg/mL的混合标准溶液,进样量为1 μL。确定各化合物的最佳质谱条件,包括离子源温度、毛细管温度、加热模块温度、加热气流速、干燥气流速、氮气流速、选择特征离子对、Q1和Q3的电压、碰撞能量等质谱分析条件。各化合物标准溶液经高效液相色谱流动相混合后注入ESI电离源,进行一级全扫描质谱分析。在正离子模式下对[M+H]+进行子离子扫描,得到主要的子离子,并选择响应值高的子离子进行碰撞能量优化,最终确定了多反应监测的离子对及碰撞能量,建立的质谱参数见表1。
3.3 提取溶剂的选择 兽药制剂基体复杂,含有大量的盐及表面活性剂。试验为避免兽药粉剂中有机盐份的干扰,选用甲醇、乙腈、0.2%甲酸乙腈3种提取溶剂对30%氟苯尼考可溶性粉阳性样品(添加浓度为0.1 mg/g)进行提取试验。选用甲醇和乙腈作为提取溶剂时,甲氧苄啶与磺胺类药物的平均回收率较高,为80.2%~99.4%,喹诺酮类药物的平均回收率为60.5%~80.2%,金刚烷胺与金刚乙胺的平均回收率为58.9%~70.6%,孔雀石绿与隐色孔雀石绿的平均回收率为66.2%~81.6%;选用0.2%甲酸乙腈作为提取溶剂时,甲氧苄啶与磺胺类药物的平均回收率为78.2%~95.2%,喹诺酮类药物的平均回收率为90.6%~101%,金刚烷胺、金刚乙胺、孔雀石绿与隐色孔雀石绿的平均回收率为83.5%~97.6%。因此本试验选用0.2%甲酸乙腈作为兽药制剂的提取溶剂。
高效液相色谱-串联质谱法应用于兽药非特定非法添加物质的检查,其检出限能达到1 μg/kg(十亿分之一),甚至更低。然而,同一车间同一流水线生产不同品种的兽药,尽管经过清场及清洗设备,还是会有少量的药物残留,一般这种残留是极低且不均匀。高效液相色谱-串联质谱法应用于兽药非特定非法添加物质的检查时更要注意选取的提取溶剂对兽药原药的提取效率,防止兽药原药对质谱造成污染,因此,经LC-MS/MS测定前用适当溶剂稀释后再上机测定。
目前,我国兽药非法添加物质检查的检测标准暂无判定限,仅要求给出检测方法的检出限或定量限。农业农村部颁布诸多公告多为高效液相色谱配二极管阵列检测法(HPLC-PDA),其检出限在1 mg/g左右。研究建立了高效液相色谱-串联质谱法同时快速测定兽药制剂中喹诺酮类、磺胺类、孔雀石绿及金刚烷胺26种药物的方法,且检出限低(0.01 mg/g)。26种药物在较宽的浓度范围内线性关系良好,该方法操作简单、重复性好,方法灵敏度高,能对兽药粉剂中常规添加药物及禁用药物进行快速筛查和准确定量。研究为兽药制剂中多种非特定添加药物快速筛查和定量提供了参考,为行政监管部门提供了可靠的技术支撑。
