具有偏向性的大麻素受体配体研究进展
2020-12-29裴方宁杨帆汤杰于丽芳
裴方宁,杨帆,汤杰,于丽芳
(华东师范大学化学与分子工程学院,上海分子治疗与新药创制工程技术研究中心,上海 200062)
G蛋白偶联受体(GPCR)属于膜蛋白家族,它由穿过质膜的7个α-螺旋构成,膜内为C端,膜外为N端。目前,靶向GPCR药物的销售额约占全球药物市场份额的27%[1]。大麻素受体属于GPCR的A家族,主要包括大麻素Ⅰ型受体(CB1)和大麻素Ⅱ型受体(CB2)2个亚型[2],此外,还有GPR18、GPR19和GPR55等[3-4]。GPCR配体包括内源性配体 anandamide(AEA)、N-花生四烯酸-多巴胺(NADA)和2-花生四烯酰甘油(2-AG)等[5-6],另外有植源性配体19-四氢大麻酚(THC)以及合成大麻素配体。目前,大麻素受体及配体存在偏向性逐渐被发现并认识,其重要性也逐渐体现。
1 大麻素受体主要亚型
CB1由472个氨基酸组成[7],主要存在于海马体、皮质、基底神经节和小脑中,是中枢神经系统中最丰富的一种GPCR[8]。CB1激动剂具有治疗疼痛、炎症以及神经退行性疾病等疾病的潜在价值,CB1拮抗剂可用于治疗肥胖以及肝纤维化等[9-11]。利莫那班(rimonabant)是首个应用于临床的CB1拮抗剂,2006年作为新型减肥药在欧盟上市,但因其存在致抑郁、焦虑甚至自杀等精神方面的严重不良反应,2008年被撤市。因此,目前研究多限于外周限制性CB1拮抗剂[12]。
CB2由360个氨基酸构成[7],与CB1的氨基酸序列存在44%同源性,跨膜区高达68%[3-4]。CB2主要存在于外周神经系统的免疫器官、组织及细胞中,在神经元中表达水平较低[2]。CB2受体与多发性硬化[13]、阿尔茨海默病[14]和肝纤维化[15]等疾病的炎症过程有关。此外,CB2受体还参与缓解疼痛、防止骨质流失等生理学过程[16]。
在CB1拮抗剂-受体复合物晶体结构中,拮抗剂以类似T形的形态结合到口袋中,CB1受体的N末端在配体识别中起关键作用:N末端的非截短部分形成V形环,插入配体结合口袋中,充当塞子以限制小分子化合物从细胞外侧进入口袋。同时,第2个细胞外环折叠成复杂结构,使氨基酸残基268 ~ 271突出到结合口袋中[2],这4种氨基酸残基对配体与受体的相互作用至关重要[17]。此外,半胱氨酸Cys257、Cys264与CB1的功能关系密切,这可能与二硫键稳定了受体的构象使得配体可识别有关(见图1)[18]。
CB1激动剂以L形结合到口袋中,相较于拮抗剂,其结构发生显著重排:螺旋Ⅰ向内弯曲6.6 Å,螺旋Ⅱ旋转约6.8 Å,螺旋Ⅵ向外移动约8Å,导致结合口袋体积减少53%。该结构重排与Phe2003.36和Trp3566.48协同构象改变关系密切,被称为“双拨动开关”。在CB1拮抗剂的结合口袋中,Phe2003.36指向远离配体结合口袋方向,并与Trp3566.48形成π-π堆积相互作用;而在CB1激动剂的结合口袋中,螺旋Ⅲ的协同旋转和Phe2003.36的侧链翻转可使苯环指向配体,同时,螺旋Ⅵ的向外旋转致Trp3566.48的侧链偏离配体,破坏了Phe2003.36和Trp3566.48的π-π堆积相互作用,因此该“双拨动开关”可能与CB1受体激活相关(见图2)[19]。
CB2拮抗剂的结合情况整体上与CB1拮抗剂不同,与其中关键残基Trp6.48构象也不同。CB2拮抗剂将Trp2586.48的侧链限制在一种相对稀有的旋转异构体中,受限的Trp2586.48构象可能会限制螺旋Ⅵ的向外移动;CB1拮抗剂将螺旋Ⅰ和螺旋Ⅱ向外推,使Phe2003.36限制Trp3566.48的移动。与CB1激动剂相似,CB2拮抗剂也以L形结合到口袋中,两者细胞外部分相似,并且关键氨基酸残基几乎相同,均包括:Phe183ECL2、Phe872.57、Val1133.32、Phe1173.36、Trp2586.48、Thr1143.33、Ile186ECL2、Trp1945.43、Ser1654.57、Phe912.61、Phe942.64和His952.65(见图 3)[20]。
2 大麻素受体信号通路
GPCR负责将细胞外信号传导至细胞质,它被特定配体激活后,与异源三聚体G蛋白结合并水解三磷酸鸟苷,从而介导下游信号传导。G蛋白由Gα、Gβ和Gγ 3个亚基组成。Gα亚基可进一步被分为 4 个亚组:Gαs、Gαi/o、Gαq/11和 Gα12/13[21]。Gα亚基可单独传导信号,而Gβ和Gγ亚基必须结合形成Gβγ才可以传导信号。GPCR偶联G蛋白后,受体活化调节关键效应物肌醇1,4,5-三磷酸(IP3)、腺苷酸环化酶(AC)等介导第二信使Ca2+、环磷酸腺苷(cAMP)等的产生。Gβγ亚基可以作为离子通道调节剂或受体激酶等调节信号传导。
GPCR不仅受G蛋白调控,还受支架蛋白的调控,受支架蛋白调控的信号通路被称为非G蛋白依赖的信号传导途径,其中研究较为充分的是β-arrestin依赖性信号传导途径[22]。GPCR活化后可释放、激活相关G蛋白,G蛋白偶联受体激酶(GRK)磷酸化GPCR细胞内的结构域,磷酸化的GPCR募集β-arrestin,使得GPCR内化和信号传导脱敏,导致传导信号的“关闭”,从而产生G蛋白依赖性信号传导的负反馈[23]。
大麻素受体主要通过抑制Gαi/o传导信号来抑制AC活性,从而导致cAMP水平降低(见图4)[24],还可以与β-arrestin作用来调控细胞外调节蛋白激酶1/2(ERK1/2)的产生[25]。在某些情况下,CB1能够与Gαs偶联,从而刺激cAMP产生,与Gαq共同调节Ca2+的产生[26],目前尚不清楚CB2是否可以与其他G蛋白结合,大麻素受体信号通路与靶标作用的关系也不明确。有研究表明,大麻素受体配体若偏向于Gαi/o信号传导通路,有利于细胞存活,保持亨廷顿蛋白突变细胞的功能,这一原理可用来治疗亨廷顿病(HD)[16];若激活Gαi/o信号传导通路,降低β-arrestin作用,可以产生镇痛作用同时降低药物成瘾性[27]。
3 偏向性信号传导
长期以来人们一直认为GPCR与G蛋白或β-arrestin偶联后可并行或有序地触发多种细胞内信号传导[28]。直到1995年,Kenakin[29]首次提出了GPCR偏向信号传导的概念。同一配体对各种细胞反应可显示出不同的功效,称为功能选择性或偏向性[30]。“功能选择性”是较早的术语,仅表示一种或多种功能的功效差异,而“偏向性”是对不同通路(例如G蛋白与β-arrestin)定量的分析[31]。偏向性信号不仅存在于G蛋白和β-arrestin之间,也存在于G蛋白不同亚型之间。目前,偏向性信号传导的具体分子机制尚不清楚,被业内普遍接受的是受体磷酸化的“条形码(BarCode)”假说。该假说认为,偏向性配体能够诱导一种独特的受体构象,然后通过效应子(如G蛋白或β-arrestin)来激活特定的信号传导途径[32-33]。例如,在第二信使蛋白Gαs存在下,β2肾上腺素受体与其激动剂的构象已被证实较为独特,TM6的胞内末端向外移动了2.1 Å,保守脯氨酸残基向外移动了1.4 Å[34]。受体构象的差别导致细胞内GRK对受体磷酸化行为的改变,进而调控β-arrestin的招募。GRK2/3和GRK5/6通过对不同C末端区域磷酸化来发挥条形码作用。GRK5/6磷酸化受体被认为是β-arrestin信号通路偏向性的标签;而GRK2/3磷酸化受体被认为是G蛋白信号通路偏向性的标签[35-36]。典型GPCR如血管紧张素I型受体(At1R)的偏向性信号研究发现,无配体结合的At1R本身就存在构象集合,大多数氨基酸残基对之间存在2种不同的距离,而不是单一的非激活态;与无配体结合相比,At1R与反向激动剂结合后,ICL2-TM6和TM5-Helix8之间的距离变化较大;At1R与激动剂结合后的变化更明显,TM1-TM6之间的距离从31 ~ 34 Å变为42 Å左右,ICL2-TM7之间的距离变短,与此同时TM7发生内倾;受体不仅在拮抗和激动时构象不同,激动不同信号通路时构象也存在差异,β-arrestin偏向性激动的受体构象中TM7内移,ICL2大幅内移,TM5小幅外移,TM6大幅外移,该构象特点会阻碍其与G蛋白偏向性配体的结合;G蛋白偏向性激动的构象特点为TM7内移,TM6外移,TM5大幅向TM6移动,ICL2基本不移动,这使得其有利于和G蛋白偏向性配体相结合[37]。该研究为偏向性信号传导机制及其他GPCR作用机制研究提供了新的思路。
目前,对GPCR偏向性配体的研究已经取得了重要进展,其中,μ-阿片受体的偏向性配体oliceridine正处于临床研究阶段。Oliceridine可以选择性地激活G蛋白通路,从而发挥镇痛作用,而不与β-arrestin通路作用[38-39],从而减轻甚至避免阿片类药物产生的便秘、呼吸抑制和成瘾性等不良反应[37]。在抗精神病药物中也发现了偏向性配体降低不良反应的情况,如aripiprazole、UNC9975和UNC9994可选择性激动β-arrestin信号通路,改善NR1基因敲除的低谷氨酸能小鼠的精神分裂症的相关症状,同时减少了与Gαi偶联相关的多巴胺依赖性过度运动等不良反应[40]。由于偏向性配体可能具有巨大的治疗优势,所以具有偏向性激活大麻素受体的药物值得大力开发。目前,对于大麻素受体偏向性的研究还处于基础阶段,首个大麻素受体偏向信号传导的证据来自Glass等[41]研究,该研究证实了CB1激动剂AEA和WIN-55212-2对Gαi激活作用比对Gαo强。短期使用CB1激动剂会产生不良的精神作用,长期服用会产生依赖性,吲哚奎尼丁酮类似物PNR-4-20和PNR-4-02是高度偏向G蛋白信号通路的激动剂,与无偏向性CB1激动剂相比,其降低了药物依赖性,减少了不良反应[42]。
量化偏向性需要同时考虑效力和最大响应,目前最常用的一种是用转导系数来衡量,即通过量化药物反应的Black-Leff模型计算转导系数τ/KA(其中τ:包含激动剂效力、受体密度和系统内的偶联;KA:解离常数,在功能测试中为亲和力的倒数)[43]。另外,还可以用相对活性(log RA)衡量偏向性,log RA = log(max/EC50)(其中max为最大响应值;EC50为激动剂效力)[44]。另外,还有比较完全激动剂和部分激动剂活性相同时的药物浓度(τ/KA)来衡量量化偏向性,此方法与相对活性一样,仅适用于浓度-响应曲线斜率无显著差异的系统[45]。
4 偏向性大麻素受体配体
4.1 CB1配体偏向性
既往研究已经证实了几种大麻素受体配体对CB1的作用存在偏向性,其不能均等地激活相关G蛋白和β-arrestin通路,因此显示出下游信号传导途径的偏向性。目前,对于CB1相关疾病的研究很多,但关于配体偏向性与其功能之间的具体联系研究较少。笔者就此总结了代表性CB1偏向性配体(内源性、植源性、大麻类似物、氨基吲哚类和二苯基类大麻素配体)及其功能(见表1)。
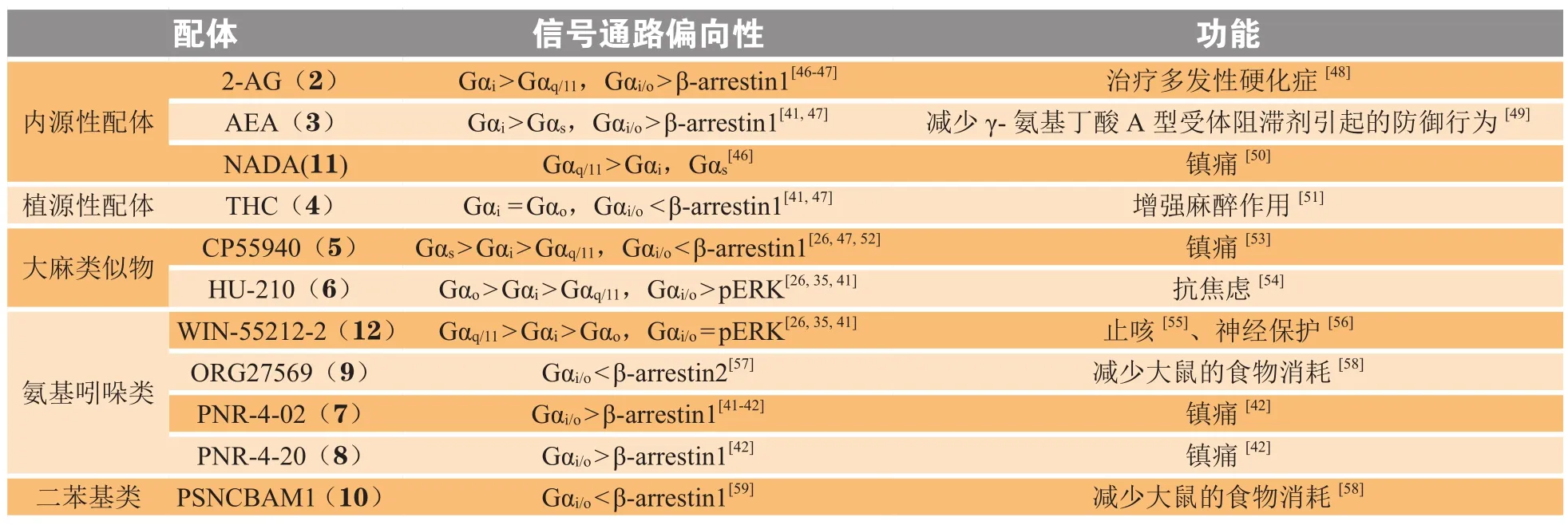
表 1 代表性CB1偏向性配体Table 1 Representative CB1 biased ligands
4.1.1 G蛋白和β-arrestin通路间偏向性 以WIN-55212(化合物1)作为对照化合物,内源性大麻素配体2-AG(化合物2)和AEA(化合物3)相对于β-arrestin1通路均优先选择Gαi/o通路并且改善HD细胞的活力;与其相反的是,植源性大麻素配体THC(化合物4)和大麻素类似物CP55940(化合物5)偏向于β-arrestin1通路并且降低HD细胞的活力。配体偏向性和细胞活力之间的关系研究表明,增强Gαi/o信号传导可能对HD治疗有益;而偏向β-arrestin1的大麻素配体可能对HD治疗不利[60]。其他合成类大麻素配体的偏向性也各有不同,例如HU-210(化合物6)优先激活Gαi/o信号,经过结构改造所得的PNR-4-02(化合物7)和PNR-4-20(化合物8)偏向于Gα信号通路并且对β-arrestin信号通路作用减弱;ORG27569(化合物9)偏向性激活β-arrestin2信号通路[41],结构优化得到的PSNCBAM1(化合物10)则优先与β-arrestin1发生作用[59]。
4.1.2 G蛋白不同亚型之间偏向性 偏向性不仅存在于G蛋白与β-arrestin之间,在G蛋白之间也存在偏向性。如2-AG(化合物2)对Gαi作用强度大于Gαq/11;AEA(化合物3)则对Gαi作用强度大于Gαs;THC(化合物4)对Gαi和Gαs作用强度相当;CP55940(化合物5)对不同亚型的G蛋白选择性存在差异,呈以下顺序 Gαs> Gαi> Gαq/11;HU-210(化合物 6)偏向于Gαi/o,与Gαq偶联较弱;NADA(化合物11)几乎只作用于Gαq信号通路以调节Ca2+,不与Gαi/o/s作用[48];WIN-55212-2(化合物 12)对 Gαi和 Gαs作用强度相当,均优先于Gαo。因此,目前绝大部分研究集中于G蛋白与β-arrestin之间的偏向性,而针对G蛋白不同亚组之间的偏向性研究较少,化合物结构特征与G蛋白亚组偏向性之间无明显对应关系。
4.2 CB2配体偏向性
CB2的配体偏向性是由Shoemaker等[61]首次发现。CB2主要与Gα蛋白偶联,CB2是否可以与其他G蛋白结合尚不清楚[52,62]。所以,对于CB2受体的研究主要是G蛋白和β-arrestin通路之间的偏向性。CP55940(化合物5)在两者间不存在明显的偏向性,以它作为对照化合物,AEA(化合物3)没有显示出偏向性;而HU910(化合物13)、HU308(化合物14)在人和小鼠的CB2呈现出不同的信号传导偏向性,它们在人CB2(hCB2)的信号转导途径表现为无偏向的平衡的激动剂,但在小鼠CB2(mCB2)上相对于β-arrestin通路,明显偏向于G蛋白信号通路[63]。因此,在测试配体时,需要考虑到种属间差异。2-AG(化合物2)、THC(化合物4)、WIN-55212-2(化合物 12)、JWH-133(化合物 15)和JWH015(化合物16)表现为G蛋白偏向性,而MH(化合物17)、AM1710(化合物18)、AM1248(化合物19)、STS-135(化合物20)和AM1241(化合物21)显示出β-arrestin偏向性质。LY2828360(化合物22)是G蛋白偏向性CB2激动剂,可减轻化疗药物诱发的神经性疼痛,而不会产生耐受性,并可以延长阿片类药物的镇痛作用时间,同时降低阿片类药物的依赖性[64]。因此,对于每个结构类型,如植源性大麻素和大麻类似物或氨基吲哚类,它们结构差异不大,但有时偏向性质却相反[65-66];或者结构发生微小的变化,其偏向性往往就会发生较大变化,这也使得对于偏向性配体的构效关系的研究变得困难(见表2)。此外,CB2配体有抗炎、镇痛等作用,相关研究也比较多,但有关偏向性配体对其功能的影响研究极少。
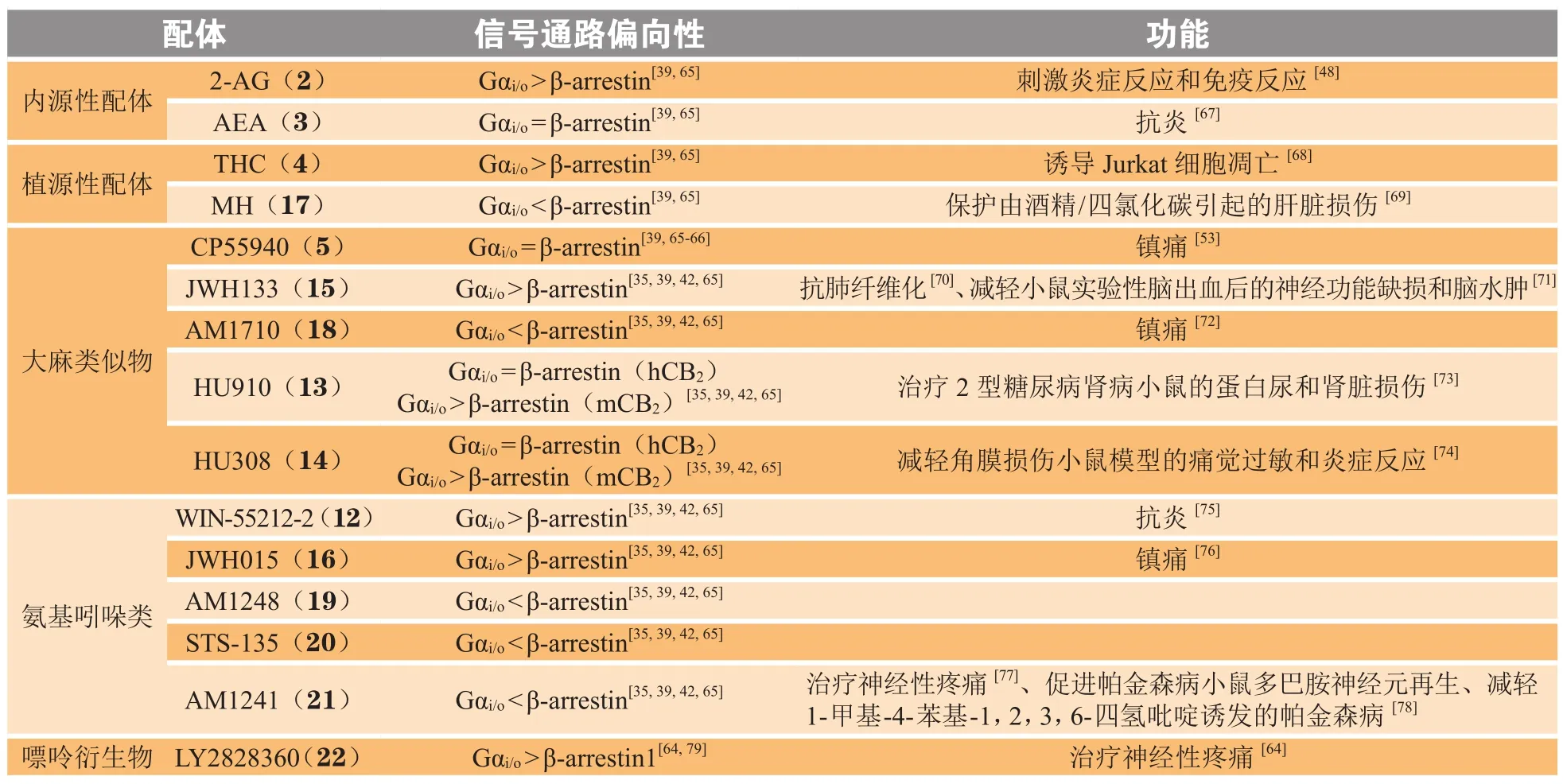
表 2 代表性CB2偏向性配体Table 2 Representative CB2 biased ligands
5 结语
综上所述,大麻素受体是疼痛、肥胖、炎症以及精神疾病的靶标,是一个非常有吸引力的研究领域。“偏向性配体”概念的提出为GPCR信号传导研究以及靶向性药物研发带来了更多方向。目前,已有证据表明配体偏向性与不良反应密切相关,例如对血管紧张素Ⅰ型受体、阿片受体等具有偏向性的配体明显降低了药物不良反应,偏向性CB2受体配体LY2828360发挥镇痛作用的同时可减少药物依赖性等,因此偏向性配体值得进一步研究。大麻素受体CB1和CB2作为GPCR家族的一员,其偏向性作用的研究尚处于初期阶段,但由于其是多效应偶联的GPCR,可介导多种生理反应,所以被广泛认为是研究偏向性信号传导的理想模型。因此,鉴定和表征具有偏向性的配体对进一步阐明大麻素受体偏向性激动和细胞内受体信号传导的机制具有重要价值。由于CB1和CB2氨基酸整体序列的同源性为44%,跨膜区序列同源性高达68%,同时,两者结合口袋关键残基高度一致,使得发展偏向性大麻素受体配体具有较高挑战性,而发展专一信号通路大麻素受体配体有利于研究信号传导机制,也有利于研究不良反应与具体信号通路的相关性,从而研发出低不良反应的大麻素配体。
