瑞博西力布的合成
2020-12-28吴童怡王文博李妍嫣
吴童怡 , 段 御 , 王文博 , 李妍嫣 , 任 畅 , 宋 磊 *
(西南民族大学 药学院,四川 成都 610225;2. 广东药科大学 药学院,广东 广州 510006)
乳腺癌是女性患病率第一位的恶性肿瘤,因乳腺癌死亡的患者数量约达 63万例[1],死亡率仅次于肺癌,在全球癌症死亡率中排名第二[2]。其中激素受体(HR)阳性/人类表皮生长因子受体(HER2)阴性乳腺癌患者占乳腺癌患者比例高达60%,针对该类患者内分泌治疗仍是首选治疗方案[3],但长期使用后会出现耐受而导致治疗失败[4],临床急需有针对性的药物。瑞博西力布(商品名Kisqali)由诺华制药研发,于2017年3月1获得FDA批准上市,是一种口服的CDK4/6选择性抑制剂[5]。该药通过阻止细胞周期从G 1期进展到S 期来防止DNA的合成,抑制肿瘤细胞增殖[6]。可以使肿瘤生长停滞时间由原先的中位数10个月提高到中位数20个月[7]。瑞博西力布被美国食品药品管理局(FDA)突破性疗法认定批准作为一线药物联合来曲唑用于一线治疗晚期或转移性HR阳性、HER2阴性乳腺癌[8]。
目前,瑞博西力布的合成路线主要有两种,一种是由诺华制药公司开发以5-溴-2,4-二氯嘧啶和环戊基胺经过亲核芳香取代反应得到5-溴-2-氯-N-环戊基嘧啶-4-胺,然后通过Sonoga-shira反应,得到3-[2-氯-4-(环戊基氨基)嘧啶-5-基]丙炔-1-醇,再在碱性条件下环化得到(2-氯-7-环戊基-7H-吡咯[2,3-d]嘧啶-6-基)甲醇,随后在MnO2以及NaCN的条件下得到2-氯-7-环戊基-N,N-二甲基-7H-吡咯[2,3-d]嘧啶-6-甲酰胺。化合物2-氯-7-环戊基-N,N-二甲基-7H-吡咯[2,3-d]嘧啶-6-甲酰胺和Boc保护的5-(哌嗪-1-基)吡啶-2-胺侧链通过Buchwald–Hartwig胺化反应得到含Boc保护基的瑞博西力布,最后在HCl存在下脱除Boc保护基,得到瑞博西力布[9]。第二种是苏州明悦医药科技有限公司开发的以N,N-二甲基-2-氧丙酰胺为起始原料经溴代后与丙二氰反应得到中间体4,4-二氰基-N,N-二甲基-2-氧代丁胺,中间体经环合得到吡咯衍生物,吡咯衍生物经烷基化反应得到氮取代化合物4-氰基-1-环戊基-5-甲氧基-N,N-二甲基-1H-吡咯-2-甲酰胺,最后氮取代化合物与 1-[5-(哌嗪-1-基)吡啶-2-基]胍环合得到目标化合物瑞博西力布[10]。上述两种方法步骤繁多,操作复杂,需要用到有毒试剂,原料不易得。
本研究开发一种新的瑞博西力布合成路线,以4-氯-2-(甲硫基)嘧啶-5-乙醛和环戊基甘氨酸乙酯为起始原料,经过环合、氧化、耦联、去保护反应得到目标产物瑞博西力布,该方法较上述两种方法而言步骤更少、收率更高、更安全环保。
1 实验
1.1 试剂与仪器
4-氯-2-(甲硫基)嘧啶-5-乙醛、环戊基甘氨酸乙酯、二环己基碳二亚胺、4-二甲氨基吡啶、间氯过氧苯甲酸等试剂从阿拉丁生化科技股份有限公司购得;一些常规溶剂如无水乙醇、石油醚、乙酸乙酯等均为分析纯,采购自天津致远化学试剂有限公司。
Varian INOVA-400核磁共振仪由美国Varian公司生产,TMS为内标;RE-52A旋转蒸发仪由上海亚荣生化仪器厂生产;SHZ-D型循环水式真空泵、TWCL-T调温磁力搅拌器由巩义市予华仪器有限责任公司生产;ZF-1型紫外分析仪由上海精密仪器仪表有限公司生产。
1.2 瑞博西力布合成路线
如图1所示,拟以化合物2(4-氯-2-(甲硫基)嘧啶-5-乙醛)和化合物3(环戊基甘氨酸乙酯)为起始原料,经过亲核取代,Aldol缩合反应合成中间体化合物5(7-环戊基-2-(甲硫基)-7H-吡咯[2,3-d]嘧啶-6-羧酸乙酯),随后中间体化合物5经过酯的氨解和氧化得到合成化合物7(环戊基-N,N-二甲基-2-(甲基磺酰基)-7H-吡咯[2,3-d]嘧啶-6-甲酰胺),化合物7最后经过耦联、去保护合成目标产物。

图1 瑞博西力布的合成路线
2 结果与讨论
2.1 化合物4的制备
向100 mL圆底烧瓶中加入1.88克化合物2(10 mmol)、1.88克化合物3(11 mmol)、50 mL无水四氢呋喃、1.01克三乙胺(10 mmol),室温下反应。TLC检测原料化合物2反应完全后停止反应,减压回收溶剂得黄色固体,石油醚/乙酸乙酯重结晶得2.80克淡黄色固体(化合物4),收率85%。1H-NMR(DMSOD6, 400 MHz)dH1.17(3H, t,J=8),1.50(2H, m),1.60~1.70(4H, m),1.94(2H, m),2.43(3H, s),4.10(2H, q,J=12,8),4.28(2H, s),4.39(2H, m),8.55(1H, s),9.75(1H, s)。MS(ESI+)m/z: 324.42[M+H]+。
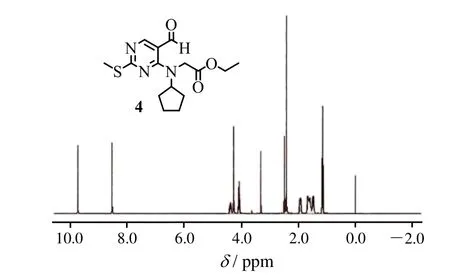
图2 化合物4的1H-NMR

图3 化合物5的1H-NMR
2.2 化合物5的制备
向50 mL圆底烧瓶中加入1.6克化合物4(5 mmol)、0.68克乙醇钠(10 mmol)、20 mL无水乙醇,回流反应,TLC检测原料消失后即停止反应,减压下回收溶剂的油状物,加水、乙酸乙酯各50 mL萃取,萃取两次合并乙酸乙酯层,减压下回收乙酸乙酯得淡黄色产物,石油醚/乙酸乙酯重结晶得1.2克化合物5,收率77%。1H-NMR(DMSO-D6, 400 MHz)dH1.34(3H, t,J=8),1.67(2H, m),2.01(4H, m),2.39(2H, m),2.57(3H, s),4.34(2H, q,J=126,8),5.72(2H, m),7.32(1H, s),8.97(1H, s)。MS(ESI+)m/z: 306.42[M+H]+。
2.3 化合物6的制备
将化合物5 0.6克(2 mmol)溶于9毫升乙醇中,加入0.53克碳酸氢钠,在室温下搅拌反应,反应完全后,用稀盐酸酸化至pH<7,乙酸乙酯萃取,萃取液用饱和盐水洗涤,无水硫酸钠干燥后浓缩得白色固体化合物,该固体化合物不经分离溶于5毫升DMF,并加入盐酸二甲胺、二环己基碳二亚胺、4-二甲氨基吡啶室温反应,反应完全后,加水和乙酸乙酯萃取,乙酸乙酯层用饱和盐水洗涤,无水硫酸钠干燥后浓缩后经得黄色产物(化合物6)0.46克。1H-NMR(DMSO-D6, 400 MHz)dH1.63(2H, m),1.96~2.02(4H,m),2.34(2H, m),2.56(3H, s),3.03(3H, s),3.05(3H, s),4.74(1H, m),6.68(1H, s),8.86(1H, s)。MS(ESI+)m/z: 305.40[M+H]+。

图4 化合物6的1H-NMR

图5 化合物7的1H-NMR
2.4 化合物7的制备
向50 mL圆底烧瓶中加入1.52克化合物6(5 mmol)、1.32克65%间氯过氧苯甲酸(10 mmol)、20 mL二氯甲烷,在室温下反应。TLC检测化合物5消失后停止反应。加水10 mL萃取两次,合并二氯甲烷层,减压下回收溶剂得黄色固体,石油醚/乙酸乙酯重结晶两次得产物(化合物7)1.1克,收率80%。1H-NMR(DMSO-D6, 400 MHz),dH1.67(2H, m),1.99~2.08(4H, m),3.01(3H, s),3.08(3H, s),3.44(3H, s),4.86(1H, m),6.96(1H, s),9.26(1H, s)。MS(ESI+)m/z: 337.42[M+H]+。
2.5 瑞博西力布(化合物1)的制备
向50 mL圆底烧瓶中加入1.68克化合物7(5 mmol)、1.84克化合物8(6 mmol)溶于35毫升甲苯,回流反应,反应完全后,减压抽干溶剂,残余物溶于30毫升甲醇,加稀盐酸室温反应,反应完全后用稀氨水碱化至pH>10,乙酸乙酯萃取后,饱和盐水洗涤,无水硫酸钠干燥后浓缩得白色固体化合物,重结晶后的目标产物(化合物1)1.4克,65%。1H-NMR(DMSO-D6, 400 MHz)dH1.63(2H,m),1.97(4H, m),2.40(2H, m),2.86(4H, m),4.73(1H, m),6.60(1H, s),7.40(1H, dd,J=9.2, 3.2),7.98(1H, d,J=2.8),8.14(1H, d,J=9.2),8.75(1H, s),9.30(1H, s)。MS(ESI+)m/z: 435.52[M+H]+。

图6 瑞博西力布化合物1的H-NMR
3 结论
本合成路线以4-氯-2-(甲硫基)嘧啶-5-乙醛和环戊基甘氨酸乙酯等简单试剂为起始原料,经过环合、氧化、耦联、去保护反应得到目标产物瑞博西力布(化合物1),合成路线所涉及到的合成策略具有原料易得、无苛刻的反应条件,各步反应收率都较高,操作简单,安全环保,有利于工业化生产,降低生产成本。以期为该药的工业化生产提供一种适宜的工艺路线。
