von-Hippel Lindau基因同义突变导致家族性嗜铬细胞瘤
2020-12-28马晓森张学斌崔云英童安莉
马晓森,张学斌,周 颋,崔云英,童安莉*
(中国医学科学院 北京协和医学院 北京协和医院 1.内分泌科; 2.泌尿外科, 北京 100730)
家族遗传性嗜铬细胞瘤/副神经节瘤(pheochromocytoma/paraganglioma, PPGL)占嗜铬细胞瘤的35%~40%,其发病与致病基因的胚系突变有关,包括VHL、RET、SDHB、SDHC和SDHD等20余种致病基因。VHL基因突变除导致家族性嗜铬细胞瘤外,临床上患者还可出现其他多种良恶性肿瘤或囊肿,包括:中枢神经系统和视网膜血管母细胞瘤、肾透明细胞癌或肾囊肿、嗜铬细胞瘤、胰腺神经内分泌肿瘤或胰腺囊肿或囊腺瘤、内淋巴囊肿瘤等[1]。由VHL基因突变导致的上述疾病称为von Hippel-Lindau (VHL) 病。VHL病为常染色体显性遗传,患病率约为1/36 000[1]。嗜铬细胞瘤在VHL病患者中发病率为10%~34%,常为VHL基因错义突变、无义突变、移码突变和大片段缺失。而同义突变仅在数个家系中进行报道[2-4]。本研究报道1个罕见的VHL同义突变导致的家族性嗜铬细胞瘤的家系,并探讨该突变在嗜铬细胞瘤中的发生频率。
1 材料与方法
1.1 材料
1.1.1 对象:先证者为2018年在北京协和医院诊治的1例双侧嗜铬细胞瘤患者,收集患者及其家系成员的临床资料及外周血。本研究获得北京协和医院伦理委员会批准(S-K431),获得了患者及其家属同意并签署知情同意书。
1.1.2 试剂盒:取先证者及其家系成员的外周血,采用Blood DNA Midi Kit (Omega Bio-Tek)提取外周血DNA。
1.2 方法
1.2.1 二代测序:将先证者外周血DNA送天津诺禾致源生物科技有限公司进行建库及全外显子测序(Illumina HiSeq PE150测序仪),并对测序结果进行生物信息学分析,采用的过滤条件如下:1) 保留测序深度>10乘位点;2) 保留千人基因组计划数据库人群等位基因频率<1 %或没有注释的位点;3) 保留外显子和剪切位点;4) 保留在“Clinvar”显示为“Pathogenic”的位点。
1.2.2 一代验证:针对二代测序发现的致病突变,采用Primer 5软件设计引物,使用聚合酶链反应(polymerase chain reaction,PCR)扩增相应外显子。扩增产物纯化后,使用荧光自动测序仪(ABI3700)直接测序。
1.2.3VHL同义突变在PPGL中发生频率:为明确致病突变在PPGL中的发生频率,在另外的107例PPGL队列中筛查是否存在该位点的胚系变异(这些样本进行了二代测序)。
2 结果
2.1 先证者及其家系成员的临床特点
先证者(Ⅳ:1,图1),男,15岁,因“阵发性心悸、大汗3月,发现双侧肾上腺占位2月”,于2018年1月就诊于北京协和医院内分泌科。患者于活动后出现发作性心悸、大汗,伴恶心和呕吐、面色苍白,发作时测血压最高达180/90 mmHg(1 mmHg=0.133 kPa)。入院检查:卧位血压:148/90 mmHg, 心率:82 beats/min;立位血压:125/85 mmHg,心率:139 beats/min,双下肢无水肿,四肢末端皮温偏凉。辅助检查:检测24 h尿儿茶酚胺显示去甲肾上腺素明显升高(表1),肾上腺增强CT显示双侧肾上腺占位(右侧占位5.1 cm×6 cm,左侧占位2.2 cm×3.8 cm),肾上腺髓质显像提示双肾上腺区占位对放射性核素摄取增高(图2),奥曲肽显像仅显示右肾上腺占位轻度放射性核素摄取,左肾上腺占位无明显摄取。患者无其他VHL病相关的临床表现及阳性检查结果。患者经过酚苄明术前准备后,于全麻下行腹腔镜双肾上腺嗜铬细胞瘤切除术。术后病理符合双肾上腺嗜铬细胞瘤,免疫组化染色:Ki-67(index2%),CgA(+),S-100(支持细胞+),α-inhibin(-),Melan-A(-),AE1/AE3(-),SDHB(+)。术后患者发作性心悸、大汗等症状消失,血压正常,复查24 h尿儿茶酚胺恢复到正常范围内。
先证者姑姥姥(Ⅱ:5)于42岁诊断“右侧肾上腺嗜铬细胞瘤”,已行手术治疗,术后15年无复发(表1),无其他VHL病相关的临床表现。否认其他家族成员患高血压或其他肿瘤。
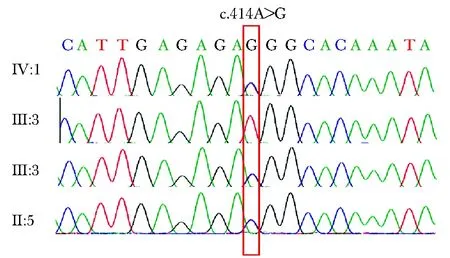
图1 VHL基因测序Fig 1 Sanger sequencing of VHL gene

图2 VHL病家系的家系图Fig 2 Pedigree of the family with VHL disease
2.2 先证者及其及家系成员的突变位点确认
先证者(Ⅳ:1)在VHL基因第二号外显子存在同义突变(c.414A>G,p.Pro138Pro,NM_000551.3),该突变在ClinVar数据库中显示为“致病性”,且在多个人群正常参考数据库中没有该位点的记录。该突变位点通过一代测序验证进行了确认(图3)。
共获得5例家系成员的胚系DNA,进行VHL基因第二号外显子的扩增及测序,结果显示,先证者母亲(Ⅲ:4)和妹妹(Ⅳ:2)均携带有该变异位点。检测这2例突变携带者的血浆甲氧基肾上腺素和甲氧基去甲肾上腺素,结果均在正常范围。
2.3 107例PPGL中该VHL同义突变的发生频率
在另外107例PPGL队列中仅发现1例患者携带有相同突变位点(c.414A>G,p.Pro138Pro)。该患者出现右侧肾上腺嗜铬细胞瘤,患者平时血压130/90 mmHg,无明显头痛、心悸、大汗等不适,检测24 h尿儿茶酚胺在正常范围,具体临床特点见表1。
3 讨论
VHL为抑癌基因, 编码的VHL蛋白, 通过与低氧诱导因子(hypoxia inducible factor,HIF)的结合使其降解,VHL突变后使VHL蛋白功能下降,则HIF-1α和HIF-2α降解减少,它们作为转录因子,促使血管内皮生长因子、促红细胞生成素和其他生长因子水平升高,从而刺激肿瘤生长。VHL基因编码产生两种不同长度的转录本:全长转录本包括外显子1、2和3(NM_000551.3)。而较短转录本仅包括外显子1和3(NM_198156.2),已知VHL蛋白与HIF结合域大部分由外显子2编码产生,因此预计较短转录本不会产生肿瘤抑制作用[5-6]。目前国外仅在8个家系[3-4,7]中检测到VHL基因同义突变(均为c.414A>G,p.Pro138Pro),功能学研究显示,该突变引起VHL剪切的改变,产生不包含外显子2的较短转录本,进而引起HIF累积,激活缺氧信号通路。由此看来,尽管同义突变因不改变氨基酸序列而被认为是沉默的突变,但仍然可以通过改变剪切位点而致病[8-10]。

A.computer tomography; B.131I-metaiodoenzylguanidine scintigraphy; C.99mTc-octreotide scintigraphy图3 先证者的影像学表现Fig 3 Radiological findings of the proband

clinical featuresⅣ:1Ⅱ:5another case with VHLsynonymous mutation∗age at onset/year154136duration/month3122highest blood pressure/mmHg180/90165/100146/90symptomspalpitation,profusesweatingnono24 h urinary catecholamine before/after operation/(μg/24 h) norepinephrine(16.69-40.65)549.64/24.21-24.64/- epinephrine(1.74-6.42)2.89/1.95-2.99/ - dopamine(120.93-330.59)280.15/181.55-226.23/ -MN(s) after operation/(nmol/L) normetanephrine(<0.9)-0.37- metanephrine (<0.5)-0.03-locationbilateral PCCright PCCright PCCtumor size/cmleft:3.0right:10.06.03.5follow up/year1152metastasisnononopresent blood pressurenormalnormalnormalfamily historyyesyesnoother clinical manifestations nonono
携带有该同义突变位点的VHL病患者,其嗜铬细胞瘤临床表现与VHL错义突变引起的嗜铬细胞瘤相似[11-12]:以去甲肾上腺素分泌为主、肿瘤常为多发性、很少恶变。对患者家系的随访,没有发现除嗜铬细胞瘤以外的VHL病相关临床表现,但是国外报道携带有此突变位点的患者中,除嗜铬细胞瘤外,还有脊髓和视网膜母细胞瘤的临床表现[2-4],这提示要对患者及其家系中突变携带者进行随访以及必要的检查,以早期发现可能的相关临床表现。
本文首次报道中国人中VHL同义突变引起VHL病的家系,该突变在PPGL队列中的发生频率约为1%。在解读VHL测序结果时要关注VHL同义突变致病的可能性,以免漏诊。
