MNase-seq与核小体定占位研究
2020-12-18邓玮杭李鑫辉
邓玮杭,李鑫辉
MNase-seq与核小体定占位研究
邓玮杭,李鑫辉
上海交通大学生物医学工程学院,上海 200240
核小体是染色质复杂三维结构的基本单位,它在基因组上的定位及占位在DNA转录、复制和修复等基础生物过程中发挥重要功能。在众多核小体定占位研究技术中,微球菌核酸酶测序(micrococcal nuclease sequencing, MNase-seq)被认为是目前最为高效的方法,因此应用十分广泛。研究人员利用该技术绘制了多种生物的核小体图谱,并揭示了核小体组织特点的共性和差异。本文介绍了MNase-seq的技术原理以及在解析核小体组织及其功能中的应用,总结了在染色质构象这一快速发展领域中的研究进展,并展望了染色质生物学的未来发展方向。由MNase-seq揭示的核小体组织结构为基因表达和发育调控提供了新的见解,也有助于人们理解疾病的发生过程。
核小体;染色质结构;染色质重塑;下一代测序技术;微球菌核酸酶
构建人类细胞的一个关键步骤是将近2 m的DNA组装成染色质并包装进直径10 μm的细胞核内,而核小体作为染色质组装的基本单位,一般是由长度为145~147 bp的DNA以左螺旋的方式缠绕组蛋白八聚体接近两圈构成的[1,2]。其中组蛋白八聚体由2个拷贝的4种核心组蛋白H2A、H2B、H3和H4组成,H1作为连接组蛋白。在基因组的大部分区域都有核小体覆盖,约75%~90%的基因组DNA包裹在核小体中[3]。核小体定位、占位以及组蛋白修饰等影响着启动子、转录起始位点的建立以及染色质高级结构的组装,参与基因转录、DNA复制和修复过程。核小体动态组织通过改变染色质结构的方式影响细胞功能,因此与生物体的基因调控、发育分化以及细胞应激过程密切相关。
应用MNase酶切配合下一代测序技术,人们加快了对核小体组织及染色质结构的研究,有望更深入了解基因调控模式以及染色质功能。本文主要介绍了微球菌核酸酶测序(micrococcal nuclease sequencing, MNase-seq)的技术原理和相关衍生技术,总结了MNase-seq等技术在揭示基因组上核小体的组织特点中的作用,综述了近年来多种生物细胞内核小体图谱的研究进展,以期为今后以核小体组织为核心的染色质构象等研究提供参考。
1 MNase-seq技术原理及其发展
1.1 MNase-seq技术原理
利用微球菌核酸酶切割染色质纤维,回收DNA并配合下一代测序技术来绘制核小体定位图谱,称作MNase-seq。尽管MNase-seq在近10年来才得以飞速发展,但早在20世纪70年代,研究人员就开始利用MNase消化染色质并研究其结构[4,5]。MNase来源于金黄色葡萄球菌(),同时具有核酸内切酶与外切酶活性。核小体间的连接DNA(linker DNA)对MNase的敏感性要比核小体DNA高25倍[6],根据酶的这一特性,在对染色质DNA进行充分消化后,大部分核小体间的裸露DNA都被消化,由此可以富集到单核小体DNA片段。
MNase-seq的主要步骤包括:(1)提取细胞核;(2)用MNase对染色质进行酶切;(3)终止酶切、去除RNA及蛋白杂质;(4)分离提纯DNA片段;(5)文库构建;(6)上机测序。在MNase-seq的实验设计中,染色质交联程度以及MNase的酶切水平很关键。不少实验利用甲醛做交联剂(crosslinking agent),在细胞活体条件下对蛋白质-DNA相互作用进行固定,以免在染色质制备过程中丢失这些相互作用及破坏核小体组织结构[7]。也有研究者用MNase直接处理未固定的染色质[8~10],该方法减少了组蛋白以外的蛋白质-DNA相互作用带来的实验误差。尽管该方法可能导致核小体结构的改变,但研究发现是否使用甲醛固定对核小体的组织改变不大[11]。使用中等或较高水平的酶切使得酶切产物中80%~100%为单核小体片段,有利于减少建库过程中片段筛选带来的额外技术误差。MNase酶切后进行电泳分析,150 bp左右的条带则是单个的完整核小体DNA。
MNase-seq的优点在于技术难度较低,具有较高的分辨率,且数据处理相对简单。较高的分辨率得益于MNase的酶切特性,MNase处理染色质可以高效去除连接DNA,得到的DNA片段末端正是包裹组蛋白的DNA两端。相比用超声打断DNA的常规方法,MNase处理DNA可以获得长度较均一的DNA片段,从而得到核小体更为精确的位置坐标。然而,由于不同DNA片段对MNase酶切的敏感性不同,这一酶切效率的差异使得MNase-seq测序片段的末端不能准确反映核小体边缘的位置,因此测序数据的处理分析尤为关键。MNase-seq数据处理过程主要为数据预处理及质量控制、序列比对、核小体定位与占位分析以及数据的可视化[12]。在序列比对后,研究者们先后运用多种算法(如iNPS[13]、DiNuP[14]等)分析MNase-seq测序数据,解析全基因组核小体定位精确图谱或对差异化占位核小体进行分析。其中,Chen等[13]建立了iNPS算法,该算法在NPS(nucleosome positioning from sequencing)的基础上增加了“核小体边界信号调整”与“相邻核小体合并或分离”步骤,它比通用的NPS算法识别核小体边界信号的能力更强,因此可以多检测到约60%的核小体。该算法具有更高的检测准确性和稳健性,因此有利于下游数据分析。核小体定位的理论预测同样具有较好的研究前景,研究者们利用核小体DNA/连接DNA的序列特性、碱基二联体周期信号等建立数学模型(如Segal模型[3]、N-Score模型[15]等)对核小体定位进行预测。近年来,MNase-seq测序数据分析方法的进步使得该技术日趋成熟,模型的改进与优化使得人们对核小体定位预测的准确性不断提高,MNase-seq技术目前已广泛应用于各种研究场景。
MNase-seq具有的这些优点使其成为检测核小体分布的优越的方法。目前,MNase-seq已被应用于酿酒酵母()、果蝇()、人类()以及多种模式生物体内的染色质结构研究。另一方面,通过MNase-seq测量染色质被核小体及其他调控因子的占有水平,可以间接揭示染色质可及性、发现潜在基因调控位点[7,12]。
1.2 MNase-seq衍生技术及相关技术
目前,以MNase-seq技术为基础,发展出了一系列衍生技术(图1),例如(1) MNase-ChIP-seq[16],被用于对特定调控因子、组蛋白修饰或变体的检测;(2) MNase-Exo-seq,在酶切体系中加入核酸外切酶Ⅲ来弥补MNase的外切酶活性,从而更高程度地切割到核小体核心区域(core particle),以获得更精准的核小体定位[17];(3) MACC-seq (MNase accessibility sequencing),同时对整个基因组上的核小体位置及
其可及性进行测量,探究核小体占位与染色质可及性的关系[18];(4) MH-seq,识别MNase高敏位点(MNase hypersensitive sites, MHSs),利用MNase可以检测到DNase I或Tn5无法访问到的开放染色质区域[19];(5) Array-seq,使用低浓度MNase酶切,用于检测核小体阵列的规律性(regularity)及核小体间距(nucleosome spacing)[20];(6) CUT&RUN (cleavage under targets and release using nuclease)技术,MNase被靶向作用于特定的组蛋白修饰位点或转录因子等蛋白结合位点[21]。
除MNase-seq及其衍生技术以外,染色质免疫共沉淀测序技术(chromatin immunoprecipitation sequencing, ChIP-seq)、染色质开放性测序技术(assay for transposase-accessible chromatin with high throughput sequencing, ATAC-seq)、DNase I超敏感位点测序(DNase I hypersensitive site sequencing, DNase-seq)以及核小体占位及甲基化测序(nucleosome occupancy and methylome sequencing, NOMe-seq)等技术也在解析核小体定位及染色质结构及功能的研究中起到重要作用。表1对这些技术进行了总结比较。
1.3 少量细胞及单细胞MNase-seq技术
多年来,大量细胞MNase-seq技术研究得较为充分并得到了广泛应用,但该方法存在两个明显的缺陷:首先,在研究临床样本等珍贵样本时,收集足够多的细胞存在困难;另外,该方法只能得到细胞群体中核小体定位的平均水平,而尽管在同源细胞群体中,细胞间的染色质状态仍存在明显的异质性,因此不能反映单个细胞内真实的核小体定位状态。随着下一代测序技术的不断进步以及单细胞测序技术的发展,MNase-seq技术也更新迭代,少量细胞起始的MNase-seq及其衍生技术、单细胞MNase-seq技术[35](single-cell MNase-seq, scMNase- seq)日趋成熟,在近年来得到重要突破。少量细胞甚至单细胞起始的MNase-seq技术有助于研究胚胎发育或疾病发展中至关重要但稀少珍贵的细胞。
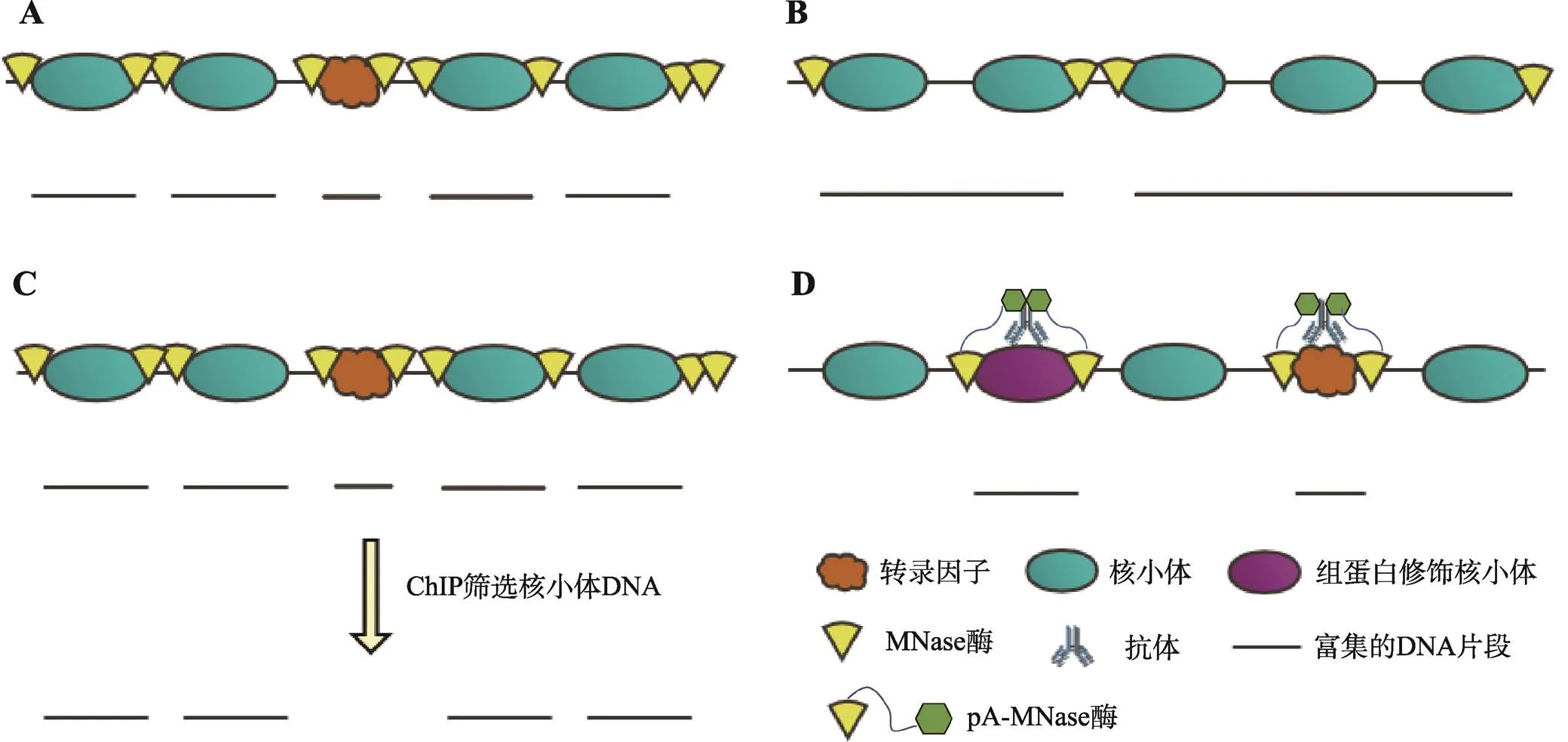
图1 MNase-seq及其衍生技术
A: MNase-seq; B: Array-seq; C: MNase-ChIP-seq; D: CUT&RUN。

表1 研究核小体、染色质结构的常用技术
Mattia等[36]发展了少量细胞MACC-seq,最低50个细胞的投入量即可达到和大量细胞MACC相似的可信结果,扩大了MACC技术的应用范围。2018年,Peter等[37]改进了CUT&RUN技术,利用该技术方法对组蛋白修饰进行测定,最低只需要投入约100个细胞。2019年,Sarah等[38]实现了在单个细胞中利用uliCUT&RUN (Ultra-low input CUT&RUN)技术对转录因子结合位点进行分析,揭示了转录因子在胚胎干细胞中结合位点的多样性。2018年,Lai等[39]开发了scMNase-seq技术,实验流程主要包括:(1)流式分选收集单个细胞;(2)裂解细胞、MNase酶切消化染色质;(3)分离提纯DNA;(4)酶切末端补平、连接测序接头、PCR扩增约25个循环;(6)筛选合适长度的片段(150~320 bp)上机测序。单个细胞含有的DNA量极少,因此scMNase-seq的技术要点在于最小化DNA损失。为达到这一目标,实验细节的优化包括在DNA提纯过程中加入环状质粒使得DNA颗粒容易可见,以及在建库流程中不预先分离单核小体长度的DNA片段。他们利用该技术同时对NIH3T3细胞、小鼠()胚胎干细胞及小鼠幼稚CD4+幼稚T细胞的全基因组核小体定位及染色质可及性进行研究。相比于大量细胞MNase-seq对核小体间距的研究只限于核小体定位精准的区域(例如TSS附近),scMNase-seq技术可以对单个细胞全基因组范围的核小体间距模式进行测量[40]。该研究表明,在异染色质区域,相邻核小体以约180 bp的间距规律排列,但在不同细胞中核小体的定位不一致;在活跃染色质区域,相邻核小体间的间隔存在差异,然而核小体的定位在不同细胞中趋于一致;在TH1增强子处,不同CD4+幼稚T细胞显示不同的核小体缺失程度,在TH1增强子处缺失程度高的细胞具有更高TH1分化的潜能。
2 MNase-seq技术解析核小体组织特点及内在功能
2.1 核小体组织特点及影响因素
核小体覆盖了可测序基因组的大部分区域,整体而言,核小体重复地、有规律地出现在基因组上。酿酒酵母基因组中,核小体以约165 bp的间隔重复出现[41],相邻核小体由10~50 bp的连接DNA(linker DNA)相连[42~45]。然而,核小体在基因组上的组织具有异质性,导致不同的染色质折叠方式。核小体在异染色质区域覆盖率高,形成了紧凑封闭的染色质结构;然而在增强子、绝缘子等调控区域常表现为核小体缺失,形成开放可及的染色质[41]。并且,核小体在基因组上的组织不是固定不变的,具有定位和结构上的动态性[46]。核小体定位的动态性体现在核小体可以在DNA上滑动,且容易自发发生全部或部分解聚的动态变化。在生理条件下,核小体易受染色质重塑复合物(chromatin remodelers)、分子伴侣、聚合酶、转录因子等调控因子的影响而发生重定位;全局转录水平的变化以及外界环境刺激也会影响核小体的定位。核小体结构的动态性体现在组蛋白的翻译后修饰(post-translational modifications, PTM)[47,48],以及组蛋白变体[49,50]和非标准核小体的形成[51]等。
研究人员使用核小体定位(nucleosome positionning)及占位(nucleosome occupancy)来描述核小体的组织状态。核小体定位指核小体出现在基因组特定位置相对于其周边的概率,反映核小体对特定DNA序列选择的特性;核小体占位是指在基因组特定区域出现的核小体平均数目,体现了核小体密度。核小体的定位和占位具有碱基偏好性,通常,富含G、C碱基的DNA序列更有利于DNA与核小体的紧密结合,而高A、T含量DNA序列削弱了DNA与组蛋白的相互作用,连续出现的A碱基区域(AAAAA)在体内和体外实验中都表现出最低的核小体占有率[52]。Albert等[53]利用MNase-ChIP-seq研究酿酒酵母核小体组织时发现并定义了DNA序列影响核小体定位的旋转特性(rotational setting)和平移特性(translational setting)。其中旋转特性与规律排列的二核苷酸有关,以10 bp为周期连续出现的AA/AT/ TA/TT二核苷酸以及相位相差5 bp的GG/CC/GC/ CG二核苷酸序列交错出现,塑造了DNA的急剧弯曲特性,有利于DNA对组蛋白八聚体的缠绕,从而具有强烈的核小体定位特性。
目前普遍认为DNA序列对于核小体在基因组上的组织起到主要作用,仅通过核小体对DNA的序列偏好可以解释体内核小体组织形态的50%~60%[3]。Kaplan等[52]利用MNase-seq对培养于3种不同培养基的酿酒酵母体内的核小体进行测定,它们的核小体图谱显示出较高的相似性,表明尽管存在环境差异,DNA序列仍是影响核小体组织的最关键因素。根据核小体定位的序列偏好性,研究人员根据DNA序列及体外合成的核小体数据建立计算模型,对生理条件下核小体组织形式进行预测[3,54]。在基因组上不同区域,DNA序列与核小体占位的相关性不同。在启动子区域,DNA序列与体内核小体占位的相关系数较非启动子区域更低。由此说明,除了DNA序列的作用,核小体在生物体内的组织形态受到众多细胞内调控因子及转录水平的影响。
以染色质重塑复合物为主的众多染色质调节因子也影响着核小体在基因组上的定位。染色质重塑复合物是一种依赖ATP的酶类,它们可以越过核小体对DNA序列的内在偏好性,利用ATP水解的能量移除、移动或并入组蛋白来改变核小体的定位及构象。在促进转录因子与DNA的结合[55]、顺式作用元件构象建成及DNA复制、转录激活等过程中发挥重要作用[56]。SWI/SNF (switch/sucrose non- fermenting)复合物是研究得较为全面的染色质重塑复合物,它们通常富集在转录起始位点、复制起点中核小体缺失位点(nucleosome-free region, NFR)的–1核小体处[57],主要通过促进核小体在DNA上的移动或移除来发挥功能,参与酿酒酵母的应激反应[58]。近年来,SWI/SNF的作用机制被研究得更为透彻,研究表明,在黑色素细胞的分化中,SWI/SNF亚基BAF60A促进色素基因及启动子对SWI/ SNF亚基BRG1的募集,从而促进染色质重塑与细胞分化[59]。ISWI(imitation SWI)复合物属于SWI蛋白家族,它能协助染色质的组装和组织[60,61],帮助具有规律间隔的核小体序列的建成[62]。CHD (chromodomain-helicase-DNA-binding)也属于SWI蛋白家族,它与ISWI共同决定了基因组上核小体间距(nucleosome spacing)的全局特征[63]。
2.2 核小体缺失位点的结构和功能
在利用下一代测序技术研究核小体组织之前,研究者们利用MNase切割染色质纤维并配合基因芯片技术(MNase-chip),在酿酒酵母的核小体组织上有了开创性的发现,即核小体在启动子区域普遍显示出缺失特性[64~67],这些区域被命名为核小体缺失位点。NFR也被发现存在于活跃增强子区域、复制起点以及转录因子结合位点[67]。核小体的缺失增加了该位点的染色质可及性、具有更强的调控潜力,利于众多反式作用因子正确发挥功能,包括染色质调控因子、转录因子、复制和转录需要的酶类等[68]。
根据染色质的可及性可将启动子分为两种类型:开放启动子(open promoter)和封闭启动子(covered promoter)[62]。开放启动子具有开放的染色质状态,在起始密码子上游200 bp左右具有一段NFR,NFR内具有暴露的转录激活因子结合位点;而封闭启动子具有较高的核小体占位,转录激活因子需要在染色质重塑复合物的帮助下与核小体竞争结合位点才能开始转录。NFR区域上下游出现的第一个核小体被命名为–1/+1核小体,分别标定了NFR的上游/下游边界[46]。+1核小体定位在转录起始位点(transcription start site, TSS)下游的固定距离,它所结合的DNA序列具有很强的核小体定位特性。利用MNase- seq对不同物种启动子的核小体结构进行测量时发现,+1核小体的位置具有物种特异性。在酵母中,+1核小体与TSS在位置上存在重叠[69],而在果蝇的基因启动子处,+1核小体通常位于TSS下游约135 bp处,因此具有比酵母更长的NFR[42](图2)。在表达基因TSS的+1核小体下游方向,形成了规律间隔、定位精准且相位统一的核小体阵列(nucleosome phasing),这一阵列大约向基因内部延伸1000 bp左右,规律排列的特性随着与TSS间距离的增大而减弱[46]。Dustin等[44]在人类CD4+T淋巴细胞中发现,+1核小体下游相位统一的核小体阵列在不表达的基因中不存在。并且,将启动子区域核小体排列与RNA聚合酶Ⅱ的ChIP-seq数据比对发现,启动子区域RNA聚合酶II水平越高,+1核小体及其下游的核小体定相现象越明显;不同种类的RNA聚合酶II影响着+1核小体的位置。

图2 酵母和果蝇+1/−1核小体的位置
A:酿酒酵母+1/−1核小体位置;B:果蝇+1/−1核小体位置。
启动子区域NFR的功能主要体现在两个方面:首先,转录起始位点的NFR有助于转录起始前复合物的组装从而开始转录,而转录终止位点附近的NFR有助于转录复合物的解聚;另外,NFR下游+1核小体与DNA的解聚或结构变化促进了RNA聚合酶Ⅱ与DNA的结合,促进了转录的进行。+1核小体的位置在转录活动中发挥重要作用,其位置向NFR上游偏移将会影响转录元件的组装,降低转录效率[70,71]。在启动子区域以外,也发现有NFR及相位统一的核小体阵列的存在,例如绝缘子复合物及转录因子的结合位点。绝缘子CTCF(CCCTC-binding factor) 蛋白结合位点处核小体占位很低,表现出核小体缺失,并且在CTCF结合位点上下游发现具有对称且规律的相位统一的核小体阵列[72]。与+1核小体类似,这些蛋白质与DNA结合很紧密,具有强定位特性,但基因组上核小体定相排布的机制还未完全破解[41]。
2.3 核小体组织与基因表达调控
核小体对DNA的包装,一方面压缩了DNA,阻碍了众多DNA结合蛋白与DNA的相互作用;另一方面,在这一过程中核小体能调整染色质的包装方式,促进了细胞内基因的正确表达[62]。核小体的定位与转录因子的结合以及基因的转录水平密切相关,且相互影响。
转录因子与核小体竞争DNA结合位点会导致核小体组织的变化,进而重塑该位点的染色质可及性状态从而调控基因表达。对于大多数转录因子而言,其对应的结合位点上有核小体存在时,由于空间位阻、电荷相斥等影响,转录因子与DNA的亲和性通常比裸露DNA低10倍以上。然而结构蛋白(architectural proteins, AP)和染色质重塑复合物能帮助转录因子与DNA的结合,这一过程通常促进核小体从DNA上分离。Daniel等[10]利用ChIP-seq结合MNase-seq等技术对35种转录因子结合位点附近的核小体组织形态进行分析,他们发现在转录因子结合位点峰值处通常形成NFR,且转录因子占位水平与其结合位点上下游核小体定位强度负相关。转录因子结合使得核小体解聚,进一步促进开放染色质的形成从而激活转录。酿酒酵母基因启动子是研究染色质结构对基因表达影响的重要模型。研究发现,在转录抑制状态的启动子处,核小体呈规律排列形式,而转录因子Pho4p与UASp2位点的结合能导致核小体组织重构,表现为–2和–3核小体解聚,核小体占位水平很低。这一核小体重构导致了染色质开放位点的形成,进而激活了PHO5的表达[73,74]。转录因子还可以通过改变局部染色质的空间结构来影响转录。基因具有两个重要的调控元件boxA与boxB,转录因子TFⅢC与这两个调控元件的结合使boxA与boxB间的核小体向上游平移约40 bp。而后,转录因子TFⅢB的结合导致–1核小体重定位从而改变TATA上游染色质空间结构,即在TATA框处形成一段对核酸酶高敏的活跃染色质,从而为转录起始前复合物(pre-initiation complex, PIC)的组装和转录做好了准备[75]。
转录水平同样也会对核小体在基因组上的定位和占位水平产生影响。Sushma等[76]利用MNase-seq对热激(heat shock)前后酿酒酵母全基因组核小体定位及占位进行测量,发现细胞在经历转录干扰(transcriptional perturbation)前后,不发生全局范围的核小体定位变化,大部分核小体的位置保持稳定,染色质重塑活动通常只与基因启动子区域单个或两个核小体的缺失或变换相关。在人CD4+T细胞中,Anton等[72]发现在不同转录频率的基因启动子处,其NFR的缺失程度不同,高表达基因(>8 RPKM)较低表达基因(<1 RPKM)的核小体缺失程度更高,因此转录活性可能与NFR区域核小体移除相关。转录活动还会影响核小体间隔,Lai等[35]利用单细胞MNase-seq技术,对小鼠CD4+T淋巴细胞和胚胎干细胞的核小体组织及染色质可及性进行研究,发现活跃转录基因内部核小体间的间隔不均匀;而在沉默基因或异染色质区域,由于没有转录活动的影响,核小体间隔高度均匀,连接DNA长度变化不大。
核小体在基因组上的排列多起到抑制转录的作用,通常与影响RNA聚合酶II的延伸及PIC的组装有关。转录过程中,核小体的存在成为了RNA聚合酶Ⅱ的延伸的物理屏障,在体内和体外的实验中都发现了核小体参与RNA聚合酶II延伸暂停(pausing process),从而影响转录效率[42,77];基因启动子区域的核小体组织特别是+1核小体的定位直接决定了PIC是否可以成功组装。Reja等[78]利用ChIP-exo技术研究酵母核糖体蛋白启动子时发现,经热激处理后,+1核小体向上游启动子区域移动导致了转录抑制,因为+1核小体对上游DNA序列的占领阻碍了PIC与足够DNA底物的结合。然而,核小体对转录的抑制作用可以被细胞内多种调控因子逆转,在人IFN-β基因启动子处,+1核小体对TATA框(TATA box)及TSS的占位阻碍了PIC的组装,抑制基因转录。当机体受到病毒感染,在增强体(enhanceosome)及其招募的SWI/SNF、GCN5乙酰转移酶等多种复合物的共同作用下,+1核小体向下游移动约36 bp,使得转录因子TFⅡD与核心启动子区域结合,从而起始转录过程[79~81]。
2.4 组蛋白修饰图谱与表观遗传学
核小体不仅是染色质组装的基本单位,也是生物体表观遗传学修饰的主要载体。核小体的定位及组蛋白翻译后修饰是表观遗传学在各领域研究中的重要内容。其中,MNase-ChIP-seq技术发挥了重要功能,该技术目前已经可以对全基因组范围内的组蛋白PTM进行捕获并测量。
组蛋白PTM被研究者们称作“组蛋白密码”(histone code),因为PTM可以直接改变染色质的物理结构,或被细胞内特定的蛋白质识别,进而激活或抑制下游染色质功能[82]。组蛋白翻译后修饰中较普遍的有赖氨酸的乙酰化、甲基化及泛素化,精氨酸的甲基化以及丝氨酸的磷酸化[83,84],这些修饰多出现在转录调控元件如启动子、增强子处。作为一种常见的修饰形式,组蛋白乙酰化削弱了组蛋白-DNA的相互作用、提高核小体周转率和染色质可及性,利于转录因子的结合从而促进基因表达,在高转录活性的基因启动子区域丰度较高[85~87]。研究表明,启动子区域H3K9ac修饰水平与基因表达水平正相关,且H3K9ac被证实在促进人胚胎干细胞分化[88]、胰岛β细胞增殖[89]等过程中起关键作用。相比于乙酰化修饰,组蛋白甲基化修饰形式更加稳定。一些甲基化形式能促进转录,如H3K4me1/3、H3K36me3等,其中H3K4me3主要分布于活跃转录基因启动子处,它有助于启动子对转录因子TFⅡD、RNA聚合酶Ⅱ的招募;在增强子处,多出现明显的H3K4me1修饰,而不出现H3K4me3修饰[90]。另一些组蛋白甲基化与基因沉默相关,H3K9me3、H3K27me3在哺乳动物细胞中促进染色质凝集[91]。
早期的研究方向多集中于H2A.Z核小体变体定位[53]与组蛋白甲基化分布形式[92]。H2A.Z作为常见的核小体变体,H2A.Z参与转录调控、DNA修复等众多生物过程[93]。H2A.Z通常出现在基因启动子处+1/−1核小体处,专一定位在+1核小体处的H2A.Z与转录起始相关[94]。H2A.Z的占位水平由它的并入水平与解离水平共同决定。其中,SWR1促进了H2A.Z的并入,而RNA聚合酶II以及Kin28/Cdk7 激酶参与H2A.Z的解离,目前,已经可以在活细胞中以单分子的分辨率追踪核小体的变化过程[95]。另一方面,H2A.Z占位水平通常与DNA甲基化水平负相关,Murphy等[96]发现在斑马鱼()的配子和胚胎发育时期,H2A.Z促使DNA甲基化模式“重编程”。其中,定位在编码胚胎早期转录因子基因启动子区的H2A.Z使得启动子处于低甲基化水平从而促进基因表达,这一过程在斑马鱼胚胎发育中发挥重要作用。
组蛋白修饰状态的改变通常与基因功能改变、细胞恶性转化相关。目前,研究人员可以利用下一代测序技术探究染色质表观遗传改变,从而分析细胞健康状态。癌细胞通常具有全局范围的表观遗传异常,其中,特定的组蛋白修饰与癌症的起始和增殖紧密相关[97]。Wang等[98]发现了H3乙酰化与肿瘤细胞耐药性的关系。他们通过抑制组蛋白去乙酰化酶HDAC3的表达,促进了癌细胞基因启动子区域的H3乙酰化,从而增强基因的表达。而PD-L1有抑制T细胞活性的作用,使得癌细胞具有更强的耐药性。这一表观遗传学机制的发现有利于发现治疗肿瘤抗药能力的潜在靶点。Aman等[99]发现在肾细胞癌中,低水平的H3K4甲基化通常与晚期癌症和肿瘤转移相关,其中H3K4me3 可能是一个较好的肿瘤转移预测标志物。Liana等[100]发现H3K9ac促进了口腔粘膜癌细胞增殖的上皮间质转化(epithelial–mesenchymal transition, EMT),因此H3K9ac可以作为口腔粘膜癌预后评估的关键标志物。表观遗传通常是动态可逆的,因此根据组蛋白表观遗传相关酶类设计药物,可以恢复“正常的表观状态”从而起到治疗效果,以组蛋白甲基转移酶抑制剂、组蛋白去乙酰化酶抑制剂为主的表观遗传学药物发展前景较为乐观[97]。
3 结语与展望
21世纪以来,下一代测序技术的高速发展加快了研究人员对染色质结构的研究,使人们在时间和空间上对核小体组织有了更清晰的认知。MNase-seq及其衍生技术揭示了全基因组范围核小体的组织方式,即核小体在基因组上的分布是动态且不均匀的,在特定区域会出现NFR。人们已经认识到核小体定位主要受到DNA序列的影响,染色质重塑因子、转录因子、聚合酶等同样参与了核小体组织及染色质结构的形成。染色质构象的建立及动态平衡使得调控元件高效地对转录因子进行招募、调控远端序列与靶基因的互作,因此与染色质功能特别是基因表达调控密切相关。
近年来,对MNase-seq技术改良的重点在于减少必要的细胞投入量及降低背景信号,目前该技术已经可以在单个细胞上实现[35],有助于人们从单个细胞的核小体组织结构层面理解细胞间的染色质异质性。在疾病研究中,将scMNase-seq与scDNase-seq、scChIP-seq以及多种表观遗传学分析方法相结合,有助于人们理解肿瘤异质性、剖析肿瘤形成过程内在的染色质结构基础、分析不同疾病的表观遗传标记,从而解决更多疾病机理问题[101]。利用MNase-seq及相关技术对全基因组核小体定位和可及性的测量初步解析了染色质结构和基因调控机制,最近发现MNase-seq还可以预测高阶染色质结构,然而其在染色质三维相互作用的分析中存在困难。将MNase- seq技术与近年来发展迅速的Hi-C[102,103]等染色质捕获技术相结合,有助于人们更好地理解染色质结构与其功能的关系。随着技术方法的不断进步以及多学科的交叉融合,人们将对复杂动态的染色质结构及功能研究得更加深入。
[1] Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution.,1997, 389(6648): 251–260.
[2] Zhou KD, Gaullier G, Luger K. Nucleosome structure and dynamics are coming of age.,2019, 26(1): 3–13.
[3] Segal E, Fondufe-Mittendorf Y, Chen L, Thåström A, Field Y, Moore IK, Wang JZ, Widom J. A genomic code for nucleosome positioning.,2006, 442(7104): 772–778.
[4] Noll M. Subunit structure of chromatin., 1974, 251(5472): 249–251.
[5] Lohr D, Kovacic RT, Van Holde KE. Quantitative analysis of the digestion of yeast chromatin by staphylococcal nuclease., 1977, 16(3): 463–471.
[6] Fan XC, Moqtaderi Z, Jin Y, Zhang Y, Liu XS, Struhl K. Nucleosome depletion at yeast terminators is not intrinsic and can occur by a transcriptional mechanism linked to 3’-end formation.,2010, 107(42): 17945–17950.
[7] Tsompana M, Buck MJ. Chromatin accessibility: a window into the genome., 2014, 7(1): 33.
[8] Zhang WL, Jiang JM. Application of MNase-Seq in the global mapping of nucleosome positioning in plants., 2018, 1830: 353–366.
[9] Pajoro A, Muiño JM, Angenent GC, Kaufmann K. Profiling nucleosome occupancy by MNase-seq: experimental protocol and computational analysis., 2018, 1675: 167–181.
[10] Gaffney DJ, McVicker G, Pai AA, Fondufe-Mittendorf YN, Lewellen N, Michelini K, Widom J, Gilad Y, Pitchard JKControls of nucleosome positioning in the human genome., 2012, 8(11): e1003036.
[11] Zhang ZH, Pugh BF. High-resolution genome-wide mapping of the primary structure of chromatin., 2011, 144(2): 175–186.
[12] Klein DC, Hainer SJ. Genomic methods in profiling DNA accessibility and factor localization., 2020, 28(1): 69–85.
[13] Chen WZ, Liu Y, Zhu SS, Green CD, Wei G, Han JDJ. Improved nucleosome-positioning algorithm iNPS for accurate nucleosome positioning from sequencing data., 2014, 5: 4909.
[14] Fu K, Tang QZ, Feng JX, Liu XS, Zhang Y. DiNuP: a systematic approach to identify regions of differential nucleosome positioning., 2012, 28(15): 1965–1971.
[15] Yuan GC, Liu JS. Genomic sequence is highly predictive of local nucleosome depletion., 2008, 4(1): e13.
[16] Wal M, Pugh BF. Genome-wide mapping of nucleosomepositions in yeast using high-resolution MNase ChIP-Seq., 2012, 513: 233–250.
[17] Ocampo J, Cui F, Zhurkin VB, Clark DJ. The proto-chromatosome: A fundamental subunit of chromatin?, 2016, 7(4): 382–387.
[18] Mieczkowski J, Cook A, Bowman SK, Mueller B, Alver BH, Kundu S, Deaton AM, Urban JA, Larschan E, Park PJ, Kingston RE, Tolstorukov MYMNase titration reveals differences between nucleosome occupancy and chromatin accessibility., 2016, 7: 11485.
[19] Zhao HN, Zhang WL, Zhang T, Lin Y, Hu YD, Fang C, Jiang JMGenome-wide MNase hypersensitivity assay unveils distinct classes of open chromatin associated with H3K27me3 and DNA methylation in., 2020, 21(1): 24.
[20] Baldi S, Krebs S, Blum H, Becker PB. Genome-wide measurement of local nucleosome array regularity and spacing by nanopore sequencing., 2018, 25(9): 894–901.
[21] Skene PJ, Henikoff S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites., 2017, 6: e21856.
[22] Rizzo JM, Bard JE, Buck MJ. Standardized collection of MNase-seq experiments enables unbiased dataset comparisons., 2012, 13: 15.
[23] Rizzo JM, Sinha S. Analyzing the global chromatin structure of keratinocytes by MNase-seq., 2014, 1195: 49–59.
[24] Cui KR, Zhao KJ. Genome-wide approaches to determining nucleosome occupancy in metazoans using MNase-Seq., 2012, 833: 413–419.
[25] Meyer CA, Liu XS. Identifying and mitigating bias in next-generation sequencing methods for chromatin biology., 2014, 15(11): 709–721.
[26] Park PJ. ChIP-seq: advantages and challenges of a maturing technology., 2009, 10(10): 669–680.
[27] Mardis ER. ChIP-seq: welcome to the new frontier., 2007, 4(8): 613–614.
[28] Buenrostro JD, Wu BJ, Chang HY, Greenleaf WJ. ATAC-seq: A method for assaying chromatin accessibility genome-Wide., 2015, 109: 21.29.1–21.29.9.
[29] Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position., 2013, 10(12): 1213–1218.
[30] Schep AN, Buenrostro JD, Denny SK, Schwartz K, Sherlock G, Greenleaf WJ. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions., 2015, 25(11): 1757–1770.
[31] Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E,Sheffield NC, Stergachis AB, Wang H, Vernot B, Garg K, John S, Sandstrom R, Bates D, Boatman L, Canfield TK, Diegel M, Dunn D, Ebersol AK, Frum T, Giste E, Johnson AK, Johnson EM, Kutyavin T, Lajoie B, Lee BK, Lee K, London D, Lotakis D, Neph S, Neri F, Nguyen ED, Qu H, Reynolds AP, Roach V, Safi A, Sanchez ME, Sanyal A, Shafer A, Simon JM, Song LY, Vong S, Weaver M, Yan YQ, Zhang ZC, Zhang ZZ, Lenhard B, Tewari M, Dorschner MO, Hansen RS, Navas PA, Stamatoyannopoulos G, Iyer VR, Lieb JD, Sunyaev SR, Akey JM, Sabo PJ, Kaul R, Furey TS, Dekker J, Crawford GE, Stamatoyannopoulos JA. The accessible chromatin landscape of the human genome., 2012, 489(7414): 75–82.
[32] Zhong JL, Luo KX, Winter PS, Crawford GE, Iversen ES, Hartemink AJ. Mapping nucleosome positions using DNase-seq., 2016, 26(3): 351–364.
[33] Kelly TK, Liu YP, Lay FD, Liang GN, Berman BP, Jones PA. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules., 2012, 22(12): 2497– 2506.
[34] Krebs AR, Imanci D, Hoerner L, Gaidatzis D, Burger L, Schübeler D. Genome-wide single-molecule footprinting reveals high RNA polymerase II turnover at paused promoters., 2017, 67(3): 411–422.e4.
[35] Lai BB, Gao WW, Cui KR, Xie WL, Tang QS, Jin WF, Hu GQ, Ni B, Zhao KJPrinciples of nucleosome organization revealed by single-cell micrococcal nuclease sequencing., 2018, 562(7726): 281–285.
[36] Lion M, Tolstorukov MY, Oettinger MA. Low-Input MNase accessibility of chromatin (low-input MACC)., 2019, 127(1): e91.
[37] Skene PJ, Henikoff JG, Henikoff S. Targeted in situ genome-wide profiling with high efficiency for low cell numbers., 2018, 13(5): 1006–1019.
[38] Hainer SJ, Bošković A, McCannell KN, Rando OJ, Fazzio TG. Profiling of pluripotency factors in single cells and early embryos., 2019, 177(5): 1319– 1329.e11.
[39] Gao WW, Lai BB, Ni B, Zhao KJ. Genome-wide profiling of nucleosome position and chromatin accessibility in single cells using scMNase-seq., 2020, 15(1): 68–85.
[40] Baldi S. Nucleosome positioning and spacing: from genome-wide maps to single arrays., 2019, 63(1): 5–14.
[41] Baldi S, Korber P, Becker PB. Beads on a string- nucleosome array arrangements and folding of the chromatin fiber., 2020, 27(2): 109–118.
[42] Mavrich TN, Jiang CZ, Ioshikhes IP, Li XY, Venters BJ, Zanton SJ, Tomsho LP, Qi J, Glaser RL, Schuster SC, Gilmour DS, Albert I, Pugh BFNucleosome organization in thegenome., 2008, 453(7193): 358–362.
[43] Lee W, Tillo D, Bray N, Morse RH, Davis RW, Hughes TR, Nislow CA high-resolution atlas of nucleosome occupancy in yeast., 2007, 39(10): 1235– 1244.
[44] Schones DE, Cui KR, Cuddapah S, Roh TY, Barski A, Wang ZB, Wei G, Zhao KJDynamic regulation of nucleosome positioning in the human genome., 2008, 132(5): 887–898.
[45] Valouev A, Ichikawa J, Tonthat T, Stuart J, Ranade S, Peckham H, Zeng K, Malek JA, Costa G, McKernan K, Sidow A, Fire A, Johnson SMA high-resolution, nucleosome position map ofreveals a lack of universal sequence-dictated positioning., 2008, 18(7): 1051–1063.
[46] Lai WKM, Pugh BF. Understanding nucleosome dynamics and their links to gene expression and DNA replication., 2017, 18(9): 548– 562.
[47] Martin C, Zhang Y. Mechanisms of epigenetic inheritance., 2007, 19(3): 266–272.
[48] Ruthenburg AJ, Li HT, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules., 2007, 8(12): 983–994.
[49] Ahmad K, Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly., 2002, 9(6): 1191–1200.
[50] Sarma K, Reinberg D. Histone variants meet their match., 2005, 6(2): 139–149.
[51] Ramachandran S, Zentner GE, Henikoff S. Asymmetric nucleosomes flank promoters in the budding yeast genome., 2015, 25(3): 381–390.
[52] Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, Segal EThe DNA-encoded nucleosome organization of a eukaryotic genome., 2009, 458(7236): 362–366.
[53] Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, Pugh BFTranslational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome., 2007, 446(7135): 572–576.
[54] Cui F, Chen LL, LoVerso PR, Zhurkin VB. Prediction of nucleosome rotational positioning in yeast and human genomes based on sequence-dependent DNA anisotropy., 2014, 15(1): 313.
[55] de Dieuleveult M, Yen KY, Hmitou I, Depaux A, Boussouar F, Bou Dargham D, Jounier S, Humbertclaude H, Ribierre F, Baulard C, Farrell NP, Park B, Keime C, Carrière L, Berlivet S, Gut M, Gut L, Werner M, Deleuze JF, Olaso R, Aude JC, Chantalat S, Pugh BF, Gérard MGenome-wide nucleosome specificity and function of chromatin remodellers in ES cells., 2016, 530(7588): 113–116.
[56] Ho L, Crabtree GR. Chromatin remodelling during development., 2010, 463(7280): 474–484.
[57] Dutta A, Gogol M, Kim JH, Smolle M, Venkatesh S, Gilmore J, Florens L, Washburn MP, Workman JLSwi/Snf dynamics on stress-responsive genes is governed by competitive bromodomain interactions., 2014, 28(20): 2314–2330.
[58] Ribeiro-Silva C, Vermeulen W, Lans H. SWI/SNF: Complex complexes in genome stability and cancer., 2019, 77: 87–95.
[59] Aras S, Saladi SV, Basuroy T, Marathe HG, Lorès P, de la Serna IL. BAF60A mediates interactions between the microphthalmia-associated transcription factor and the BRG1-containing SWI/SNF complex during melanocyte differentiation., 2019, 234(7): 11780–11791.
[60] Oppikofer M, Bai TY, Gan YT, Haley B, Liu P, Sandoval W, Ciferri C, Cochran AGExpansion of the ISWI chromatin remodeler family with new active complexes., 2017, 18(10): 1697–1706.
[61] Levendosky RF, Bowman GD. Asymmetry between the two acidic patches dictates the direction of nucleosome sliding by the ISWI chromatin remodeler., 2019, 8: e45472.
[62] Cairns BR. The logic of chromatin architecture and remodelling at promoters., 2009, 461(7261): 193–198.
[63] Ocampo J, Chereji RV, Eriksson PR, Clark DJ. The ISW1 and CHD1 ATP-dependent chromatin remodelers compete to set nucleosome spacing., 2016, 44(10): 4625–4635.
[64] Lee CK, Shibata Y, Rao B, Strahl BD, Lieb JD. Evidence for nucleosome depletion at active regulatory regions genome-wide., 2004, 36(8): 900–905.
[65] Sekinger EA, Moqtaderi Z, Struhl K. Intrinsic histone-DNA interactions and low nucleosome density are important for preferential accessibility of promoter regions in yeast., 2005, 18(6): 735–748.
[66] Bernstein BE, Liu CL, Humphrey EL, Perlstein EO, Schreiber SL. Global nucleosome occupancy in yeast., 2004, 5(9): R62.
[67] Yuan GC, Liu YJ, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJGenome-scale identification of nucleosome positions in., 2005, 309(5734): 626–630.
[68] Rando OJ, Ahmad K. Rules and regulation in the primary structure of chromatin., 2007, 19(3): 250–256.
[69] Mavrich TN, Ioshikhes IP, Venters BJ, Jiang CZ, Tomsho LP, Qi J, Schuster SC, Albert I, Pugh BFA barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome., 2008, 18(7): 1073–1083.
[70] Kubik S, Bruzzone MJ, Challal D, Dreos R, Mattarocci S, Bucher P, Libri D, Shore DOpposing chromatin remodelers control transcription initiation frequency and start site selection., 2019, 26(8): 744–754.
[71] Kubik S, O’Duibhir E, de Jonge WJ, Mattarocci S, Albert B, Falcone JL, Bruzzone MJ, Holstege FCP, Shore DSequence-directed action of RSC remodeler and general regulatory factors modulates +1 nucleosome position to facilitate transcription., 2018, 71(1): 89–102.e5.
[72] Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A. Determinants of nucleosome organization in primary human cells., 2011, 474(7352): 516– 520.
[73] Boeger H, Griesenbeck J, Strattan JS, Kornberg RD. Removal of promoter nucleosomes by disassembly rather than sliding., 2004, 14(5): 667–673.
[74] Ertel F, Dirac-Svejstrup AB, Hertel CB, Blaschke D, Svejstrup JQ, Korber P. In vitro reconstitution of PHO5 promoter chromatin remodeling points to a role for activator-nucleosome competition., 2010, 30(16): 4060–4076.
[75] Shivaswamy S, Bhargava P. Positioned nucleosomes due to sequential remodeling of the yeast U6 small nuclear RNA chromatin are essential for its transcriptional activation., 2006, 281(15): 10461–10472.
[76] Shivaswamy S, Bhinge A, Zhao YJ, Jones S, Hirst M, Iyer VR. Dynamic remodeling of individual nucleosomes across a eukaryotic genome in response to transcriptional perturbation., 2008, 6(3): e65.
[77] Kulaeva OI, Hsieh FK, Chang HW, Luse DS, Studitsky VM. Mechanism of transcription through a nucleosome by RNA polymerase II., 2013, 1829(1): 76–83.
[78] Reja R, Vinayachandran V, Ghosh S, Pugh BF. Molecular mechanisms of ribosomal protein gene coregulation., 2015, 29(18): 1942–1954.
[79] Lomvardas S, Thanos D. Modifying gene expression programs by altering core promoter chromatin architecture., 2002, 110(2): 261–271.
[80] Ford E, Thanos D. The transcriptional code of human IFN-beta gene expression., 2010, 1799(3–4): 328–336.
[81] Au-Yeung N, Horvath CM. Transcriptional and chromatin regulation in interferon and innate antiviral gene expression., 2018, 44: 11–17.
[82] Rothbart SB, Strahl BD. Interpreting the language of histone and DNA modifications., 2014, 1839(8): 627–643.
[83] Felsenfeld G, Groudine M. Controlling the double helix., 2003, 421(6921): 448–453.
[84] Qi HY, Zhang ZJ, Li YJ, Fang XD. Role of chromatin conformation in eukaryotic gene regulation., 2011, 33(12): 1291–1299.亓合媛, 张昭军, 李雅娟, 方向东. 染色质构象调控真核基因的表达. 遗传, 2011, 33(12): 1291–1299.
[85] Vogelauer M, Wu J, Suka N, Grunstein M. Global histone acetylation and deacetylation in yeast., 2000, 408(6811): 495–498.
[86] Bernstein BE, Humphrey EL, Erlich RL, Schneider R, Bouman P, Liu JS, Kouzarides T, Schreiber SLMethylation of histone H3 Lys 4 in coding regions of active genes., 2002, 99(13): 8695–8700.
[87] Klemm SL, Shipony Z, WJ. Chromatin accessibility and the regulatory epigenome., 2019, 20(4): 207–220.
[88] Du YH, Liu ZP, Cao XK, Chen XL, Chen ZY, Zhang XB, Zhang XQ, Jiang CZNucleosome eviction along with H3K9ac deposition enhances Sox2 binding during human neuroectodermal commitment., 2017, 24(6): 1121–1131.
[89] Daneshpajooh M, Bacos K, Bysani M, Bagge A, Ottosson Laakso E, Vikman P, Eliasson L, Mulder H, Ling CHDAC7 is overexpressed in human diabetic islets and impairs insulin secretion in rat islets and clonal beta cells., 2017, 60(1): 116–125.
[90] Vallianatos CN, Raines B, Porter RS, Bonefas KM, Wu MC, Garay PM, Collette KM, Seo YA, Dou Y, Keegan, CE, Tronson NC, Iwase SMutually suppressive roles of KMT2A and KDM5C in behaviour, neuronal structure, and histone H3K4 methylation., 2020, 3(1): 278.
[91] Sasidharan Nair V, El Salhat H, Taha RZ, John A, Ali BR, Elkord E. DNA methylation and repressive H3K9 and H3K27 trimethylation in the promoter regions of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, and PD-L1 genes in human primary breast cancer., 2018, 10: 78.
[92] Barski A, Cuddapah S, Cui KR, Roh TY, Schones DE, Wang ZB, Wei G, Chepelev L, Zhao KJHigh-resolution profiling of histone methylations in the human genome., 2007, 129(4): 823–837.
[93] Giaimo BD, Ferrante F, Herchenröther A, Hake SB, Borggrefe T. The histone variant H2A.Z in gene regulation., 2019, 12(1): 37.
[94] Bagchi DN, Battenhouse AM, Park D, Iyer VR. The histone variant H2A.Z in yeast is almost exclusively incorporated into the +1 nucleosome in the direction of transcription., 2020, 48(1): 157–170.
[95] Ranjan A, Nguyen VQ, Liu S, Wisniewski J, Kim JM, Tang XN, Mizuguchi G, Elalaoui E, Nickels TJ, Jou V, English BP, Zheng QS, Luk E, Lavis, LD, Lionnet T, Wu CLive-cell single particle imaging reveals the role of RNA polymerase II in histone H2A.Z eviction., 2020, 9: e55667.
[96] Murphy PJ, Wu SF, James CR, Wike CL, Cairns BR. Placeholder nucleosomes underlie Germline-to-Embryo DNA methylation reprogramming., 2018, 172(5): 993–1006.e13.
[97] Sharma S, Kelly TK, Jones PA. Epigenetics in cancer., 2010, 31(1): 27–36.
[98] Wang HF, Fu C, Du J, Wang HS, He R, Yin XF, Li HX, Li X, Wang HX, Li K, Zheng L, Liu ZC, Qiu YREnhanced histone H3 acetylation of the PD-L1 promoter via the COP1/c-Jun/HDAC3 axis is required for PD-L1 expression in drug-resistant cancer cells., 2020, 39(1): 29.
[99] Kumar A, Kumari N, Sharma U, Ram S, Singh SK, Kakkar N, Kaushal K, Prasad RReduction in H3K4me patterns due to aberrant expression of methyltransferases and demethylases in renal cell carcinoma: prognostic and therapeutic implications., 2019, 9(1): 8189.
[100] Webber LP, Wagner VP, Curra M, Vargas PA, Meurer L, Carrard VC, Squarize CH, Castilho RM, Martins MDHypoacetylation of acetyl-histone H3 (H3K9ac) as marker of poor prognosis in oral cancer., 2017, 71(2): 278–286.
[101] Torres CM, Biran A, Burney MJ, Patel H, Henser- Brownhill T, Cohen AHS, Li YL, Ben-Hamo R, Nye E, Spencer-Dene B, Chakravarty P, Efroni S, Matthews N, Misteli T, Meshorer E, Scaffidi PThe linker histone H1.0 generates epigenetic and functional intratumor heterogeneity., 2016, 353(6307): aaf1644.
[102] Huang QT, Li Q, Zhang YB. Linking chromatin conformation to gene function., 2020, 42(1): 1–17.黄其通, 李清, 张玉波. 染色质构象与基因功能. 遗传, 2020, 42(1): 1–17.
[103] Denker A, de Laat W. The second decade of 3C technologies: detailed insights into nuclear organization., 2016, 30(12): 1357–1382.
Resolving nucleosomal positioning and occupancy with MNase-seq
Weihang Deng, Xinhui Li
Nucleosomes are the basic unit of the three-dimensional structure of chromatin. It is now widely accepted that the positioning and occupancy of nucleosomes play important roles in fundamental genomic processes such as DNA transcription, replication and repair. Among the methods used to provide genome-wide nucleosomal positions and occupancy levels, MNase-seq has proven to be highly effective. Indeed, with this method, the nucleosomal landscapes of a variety of organisms have now been investigated, revealing both commonalities and differences. In this review, we first introduce the technical principles underlying MNase-seq, focusing on details essential to precisely resolve nucleosome positioning and occupancy. We then describe recent advances with this method, as well as future perspectives of its role in chromatin biology, with a particular focus of uncovering mechanistic insights of many disease process.
nucleosome; chromatin structure; chromatin remodeling; next-generation sequencing (NGS); micrococcal nuclease
2020-09-04;
2020-10-18
国家自然科学基金项目(编号:81972909)资助[Supported by the National Natural Science Foundation of China(No. 81972909)]
邓玮杭,在读硕士研究生,专业方向:系统生物医学。E-mail: weihangdeng@sjtu.edu.cn
李鑫辉,博士,助理研究员,研究方向:系统生物学与分子生物学。E-mail: xhli@sjtu.edu.cn
10.16288/j.yczz.20-178
2020/11/9 11:17:28
URI: https://kns.cnki.net/kcms/detail/11.1913.R.20201106.1053.003.html
(责任编委: 李海涛)
