HPLC柱前衍生法测定棉籽蛋白中17种氨基酸含量
2020-12-15杨清山程远欣刘登帅吉桂珍刘翠翠张晓芳
杨清山,程远欣,2,刘登帅,2,吉桂珍,刘翠翠,2,张晓芳
(1.晨光生物科技集团股份有限公司,河北 邯郸 057250; 2.河北省棉籽综合加工技术创新中心(筹),河北 邯郸 057250)
棉籽蛋白是一种很好的食用蛋白源和饲用蛋白源。我国每年产棉籽700万t左右,开发利用好这一资源,可以快捷、经济地解决我国饲料蛋白的不足。为确定棉籽蛋白在饲料中应用的优势及配比,需要对其氨基酸进行准确的检测分析。因此,建立一种棉籽蛋白中氨基酸的检测方法,对于推进棉籽蛋白在饲料中的应用具有重要意义。
目前,氨基酸测定方法主要包括氨基酸分析仪法[1-2]、离子色谱法[3]和高效液相色谱法[4]等,前两种方法需要专门仪器,普适性较差,而高效液相色谱法主要采用柱前衍生法[5-7]和柱后衍生法[8]。目前常用柱前衍生试剂有异硫氰酸苯酯(PITC)[9]、丹磺酰氯[10]、邻苯二甲醛-9-芴甲基氯甲酸酯(OPA-FMOC)[11]、邻苯二甲醛(OPA)[12]和6-氨基喹啉-N-羟基琥珀酰亚胺基氨基甲酸酯(AQC)[13]。柱前衍生PITC法反应速度慢且易受试剂干扰;柱前衍生OPA-FMOC法与高效液相色谱仪配套使用,易受仪器限制;柱前衍生AQC法试剂保质期短,国内购买货期长。2,4-二硝基氯苯(2, 4-dinitrochlorobenzene,CDNB)作为一种新型的氨基酸柱前衍生试剂,可克服上述缺点,并具有衍生产物性质稳定、重现性好、衍生试剂毒性小、衍生条件简单、过量的衍生试剂不干扰测定结果等优点,被广泛使用。
在综合考虑各种氨基酸检测方法优缺点的条件下,本文建立了一种酸解处理、2,4-二硝基氯苯(2, 4-dinitrochlorobenzene,CDNB)衍生、内标法定量高效液相色谱检测棉籽蛋白中17种氨基酸含量的方法。
1 材料与方法
1.1 实验材料
1.1.1 原料与试剂
棉籽蛋白,晨光生物科技集团股份有限公司。
乙腈(色谱纯,美国SR公司);乙腈、盐酸、氢氧化钠、冰乙酸、乙酸钠、三乙胺、碳酸氢钠、碳酸钠、苯酚,分析纯;2,4-二硝基氯苯(纯度大于99.0%,东京化学工业株式会社);DL-2-氨基丁酸(α-氨基丁酸)(纯度大于99.0%,梯希爱(上海)化成工业发展有限公司);17种氨基酸标准品混标(2.5 mmol/mL,Sigma公司)。
1.1.2 仪器与设备
Waters E2695高效液相色谱仪,配备2489紫外可见检测器; TH-600型超声波清洗器;岛津 AUY220型分析天平; JJ-300电子天平;DK-98-IIA型电热恒温水浴锅; FW100型万能粉碎机;101-0N电热鼓风干燥箱; R213B旋转蒸发仪。
1.2 实验方法
1.2.1 标准溶液的配制
1.2.1.1α-氨基丁酸内标溶液配制
准确称取0.2 g(精确至0.1 mg)左右α-氨基丁酸,用0.1 mol/L盐酸溶液溶解定容至100 mL容量瓶中,得到2.0 mg/mL的内标溶液。室温保存,有效期1个月。
1.2.1.2 17种氨基酸混合标准工作溶液配制
分别移取一定体积的含有17种氨基酸质量浓度约为0.3 mg/mL混标溶液至25 mL容量瓶中,加入2 mL 2.0 mg/mL的α-氨基丁酸内标溶液,使用0.1 mol/L盐酸定容,得到系列混标溶液。冷冻保存,有效期1年。
1.2.2 样品的前处理
1.2.2.1 酸解
将样品用粉碎机粉碎至40目,混合均匀。准确称取0.2 g粉碎的样品(总氨基酸含量50%)置于专用的35 mL水解管中,用移液管加入含0.1%苯酚的6 mol/L盐酸10 mL,通入高纯氮气,立即拧紧瓶盖,放入110℃电热鼓风干燥箱中酸解24 h。
1.2.2.2 转移和复溶
将1.2.2.1酸解样品取出冷却至室温,转移至25 mL容量瓶中,用0.1 mol/L盐酸清洗内壁,用移液管加入2.0 mg/mL内标溶液2 mL,最后用0.1 mol/L盐酸定容,混匀,静置约10 min。用移液管取上清液2 mL于150 mL旋蒸瓶中,在70℃电热恒温水浴锅中用真空泵抽真空(约5 min),浓缩至干,用移液枪取2 mL 0.1 mol/L的盐酸溶液复溶,超声溶解,待衍生。
1.2.2.3 衍生
使用10 mL具塞密封玻璃管,加入200 μL 样品酸解复溶液、1 mL碳酸钠缓冲盐溶液(pH 9.0)和200 μL 3% 2,4-二硝基氯苯乙腈溶液,拧紧塞子,在95℃电热恒温水浴锅中避光反应(150±10)min。
1.2.2.4 中和
将1.2.2.3的样品取出后,冷却至室温,加入200 μL 10%冰乙酸调pH为中性,超声反应排气泡,取上清液过有机膜,待高效液相色谱检测。
1.2.3 色谱条件
Waters Symmetry C18色谱柱(5 μm,4.6 mm×250 mm);柱温40℃;检测波长360 nm;进样量10 μL;流速1.5 mL/min;运行时间30 min;流动相A为乙腈,流动相B为乙酸-乙酸钠缓冲液(2.5 g乙酸钠、1.5 mL三乙胺和1.17 mL冰乙酸溶于1 L纯净水中),流动相梯度洗脱条件见表1。

表1 流动相梯洗脱度条件

续表1
1.2.4 数据处理
分别检测衍生后的混合标准工作液和样品液,积分记录峰面积,采用内标单点定量法,计算得到样品中的各氨基酸含量。
2 结果与分析
2.1 方法建立和优化
选择乙腈和乙酸-乙酸钠缓冲液作为流动相,分别使用Agilent AAA色谱柱(3.5 μm, 4.6 mm×150 mm)和Waters Symmetry C18色谱柱(5 μm,4.6 mm×250 mm),验证不同流动相梯度洗脱条件下的分离效果。结果表明:使用Agilent AAA色谱柱 (3.5 μm,4.6 mm×150 mm)时,17种氨基酸色谱峰分离度低,峰形差;使用Waters Symmetry C18色谱柱(5 μm, 4.6 mm×250 mm)时,17种氨基酸色谱峰分离度高,峰形良好,检测时间可以控制在30 min以内(见图1,标准品成分及保留时间见表2)。因此,本方法选择Waters Symmetry C18色谱柱(5 μm,4.6 mm×250 mm)。
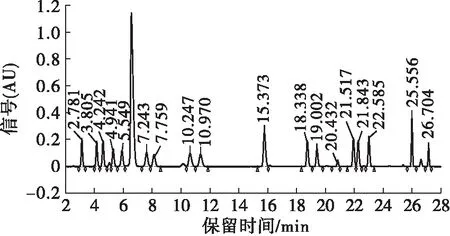
图1 17种氨基酸标准溶液色谱图
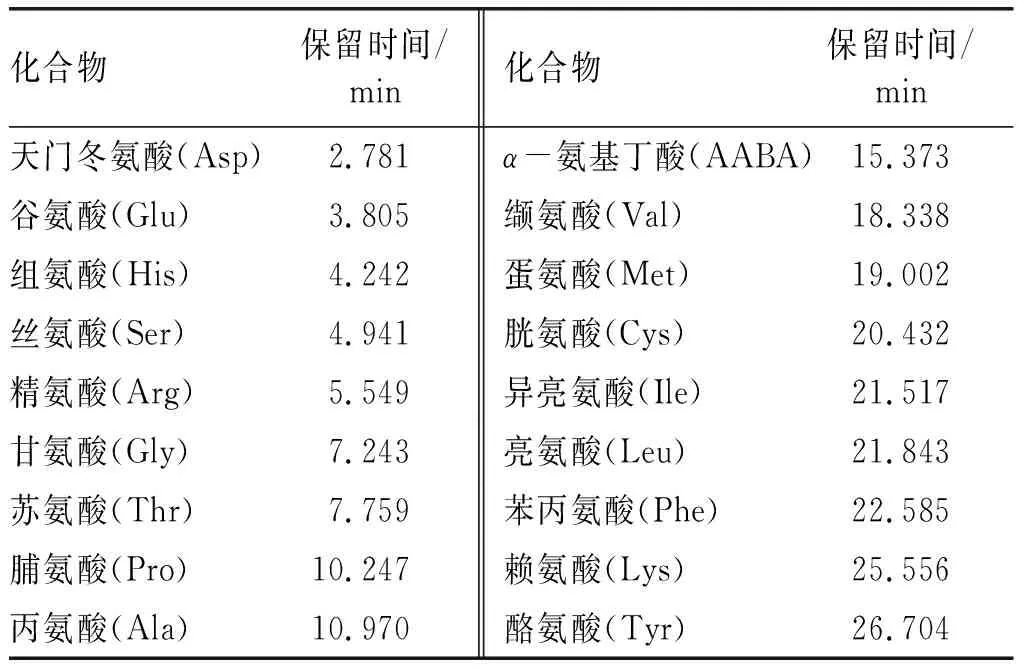
表2 标准品成分及保留时间
2.2 线性关系、检出限和定量限
按照1.2.1配制系列标准工作溶液,根据1.2.2和1.2.3方法进行衍生和检测,以分析物峰面积对被测组分的质量浓度绘制标准曲线,结果见表3。
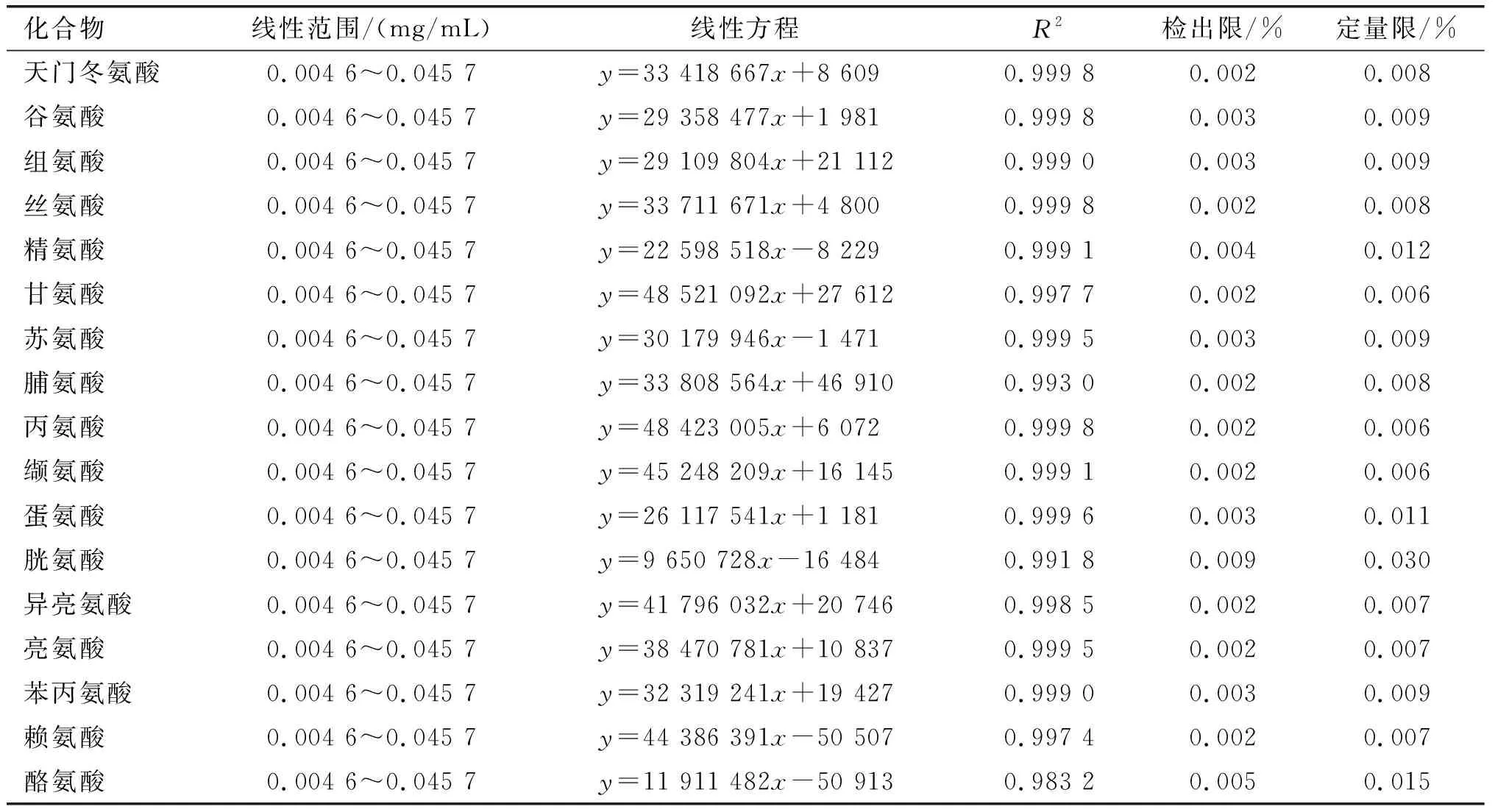
表3 方法的线性范围、线性方程、检出限和定量限
由表3可知,17种氨基酸在0.004 6~0.045 7 mg/mL范围内呈良好线性关系,相关系数(R2)为0.983 2~0.999 8。以3倍和10倍的信噪比分别确定了方法的检出限和定量限(见表3),17种氨基酸的检出限在0.002%~0.009%之间,定量限在0.006%~ 0.030%之间。
2.3 酸解方式的选择
固定前处理方法和色谱条件,选取具有外部检测结果的棉籽蛋白样品,分别按照烘箱法(110℃酸解24 h)和微波消解法(160℃酸解25 min)酸解样品,对比两种酸解方式检测结果与外部检测结果的相对偏差水平。结果表明:烘箱法酸解样品检测结果较微波消解法酸解样品检测结果与外部检测结果的相对偏差小。
选择同一批样品,每天处理3个平行样,连续处理3 d,考察微波消解法酸解样品的精密度,结果见表4。
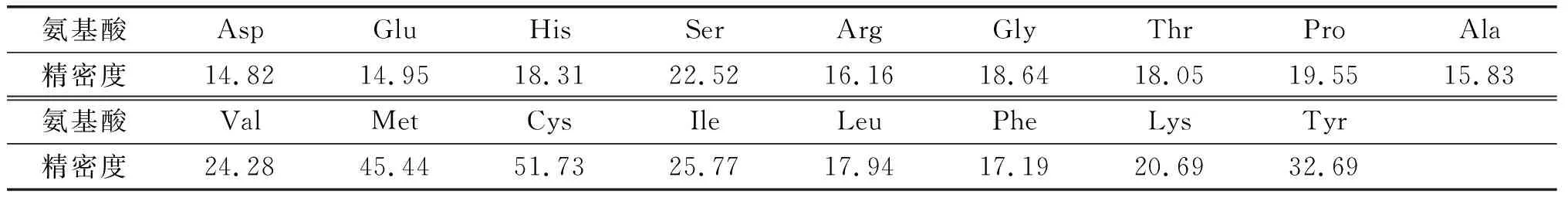
表4 微波消解法酸解精密度验证(n=9) %
由表4可知,17种氨基酸检测精密度均大于10%,最高50%左右,说明方法的精密度较差。
综上,微波消解法检测结果准确性、稳定性均较差,因此本文选择烘箱法酸解样品。
2.4 衍生参数条件优化和验证
2.4.1 不同衍生反应容器选择和验证
分别使用2 mL离心管、5 mL样品瓶(密封)、10 mL玻璃瓶(非密封)、10 mL玻璃管(密封)和20 mL顶空瓶5种容器衍生17种氨基酸标准溶液,分析峰面积响应情况。结果表明,使用10 mL玻璃管(密封)衍生反应曲线相对稳定,操作方便。因此,本文选择10 mL玻璃管(密封)作为衍生反应容器。
2.4.2 不同衍生反应体系和方式选择及验证
分别在3%的乙腈水体系90℃加热、3%的乙腈水体系95℃加热、3%的甲醇水体系95℃加热、3%的甲醇水体系75℃超声、30%的甲醇水体系90℃加热、30%的乙腈水体系75℃超声、30%的乙腈水体系90℃加热的衍生条件下,衍生不同的时间,分析峰面积响应情况。结果表明:所选取的各种衍生条件均无法达到最大值,只能达到相对的稳定值;衍生温度越高衍生效果越好;采用加热的方式衍生效果比采用超声衍生的效果好;乙腈衍生体系的衍生效果较甲醇衍生体系的衍生效果好。根据衍生反应响应曲线,本文选择衍生反应条件为:3%乙腈体系,在95℃水浴条件下衍生(150±10)min。
2.5 精密度验证
样品前处理步骤较长,包括称样、酸解、浓缩和衍生等步骤,各步骤均对检测精密度产生影响,因此本文采取倒推的方式对每步进行了精密度验证。
2.5.1 衍生步骤精密度验证
选取同一样品液,每天平行衍生3次,连续衍生3 d,考察衍生精密度,结果见表5。
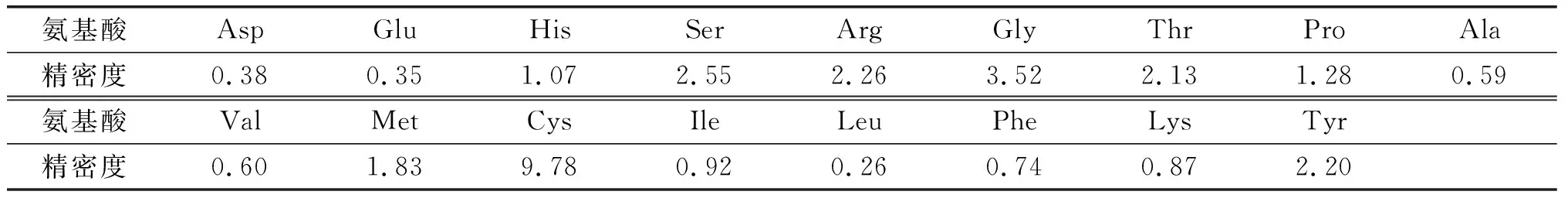
表5 衍生步骤精密度验证 %
由表5可知,除胱氨酸由于含量水平低精密度为9.78%外,其他16种氨基酸精密度均在4%以内,总氨基酸衍生步骤精密度为0.59%,说明衍生步骤精密度良好。
2.5.2 浓缩步骤精密度验证
选取同一浓缩前的样品液,日内重复浓缩5次,连续处理3 d,考察浓缩步骤精密度,结果见表6。
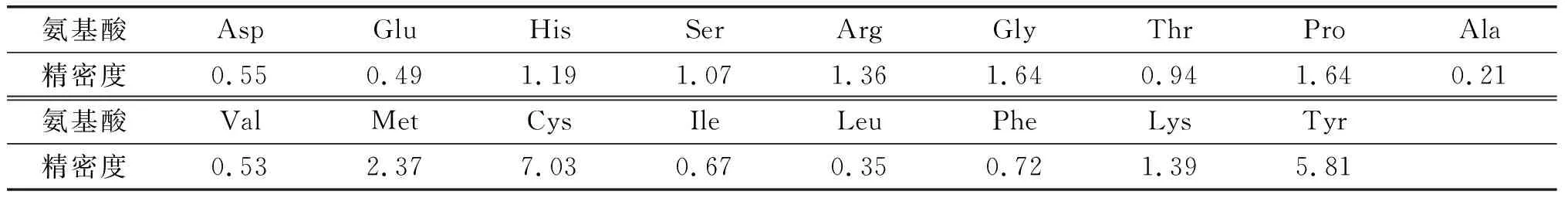
表6 浓缩步骤精密度验证 %
由表6可知,除胱氨酸和酪氨酸精密度大于5%外,其他15种氨基酸精密度均在3%以内,总氨基酸浓缩步骤精密度为0.32%,说明浓缩步骤精密度良好。
2.5.3 方法精密度验证
选取同一样品,日内重复检测5次,连续检测3 d,考察方法的精密度,结果见表7。由表7可知,蛋氨酸、胱氨酸检测精密度较差。分析原因:一是棉籽蛋白中蛋氨酸、胱氨酸含量较低;二是这两种氨基酸属于含硫氨基酸,直接酸解时稳定性差,需要使用氧化水解方式才能准确检测。其余15种氨基酸精密度在1.71%~8.43%之间。整体上方法精密度较好,总氨基酸方法精密度为1.43%。
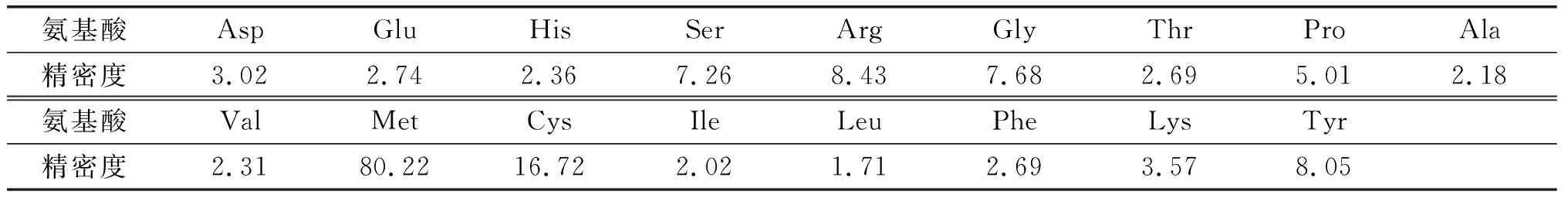
表7 方法精密度验证 %
2.6 回收率
在3个水平下进行样品加标回收率实验,考察方法的准确度,结果见表8。
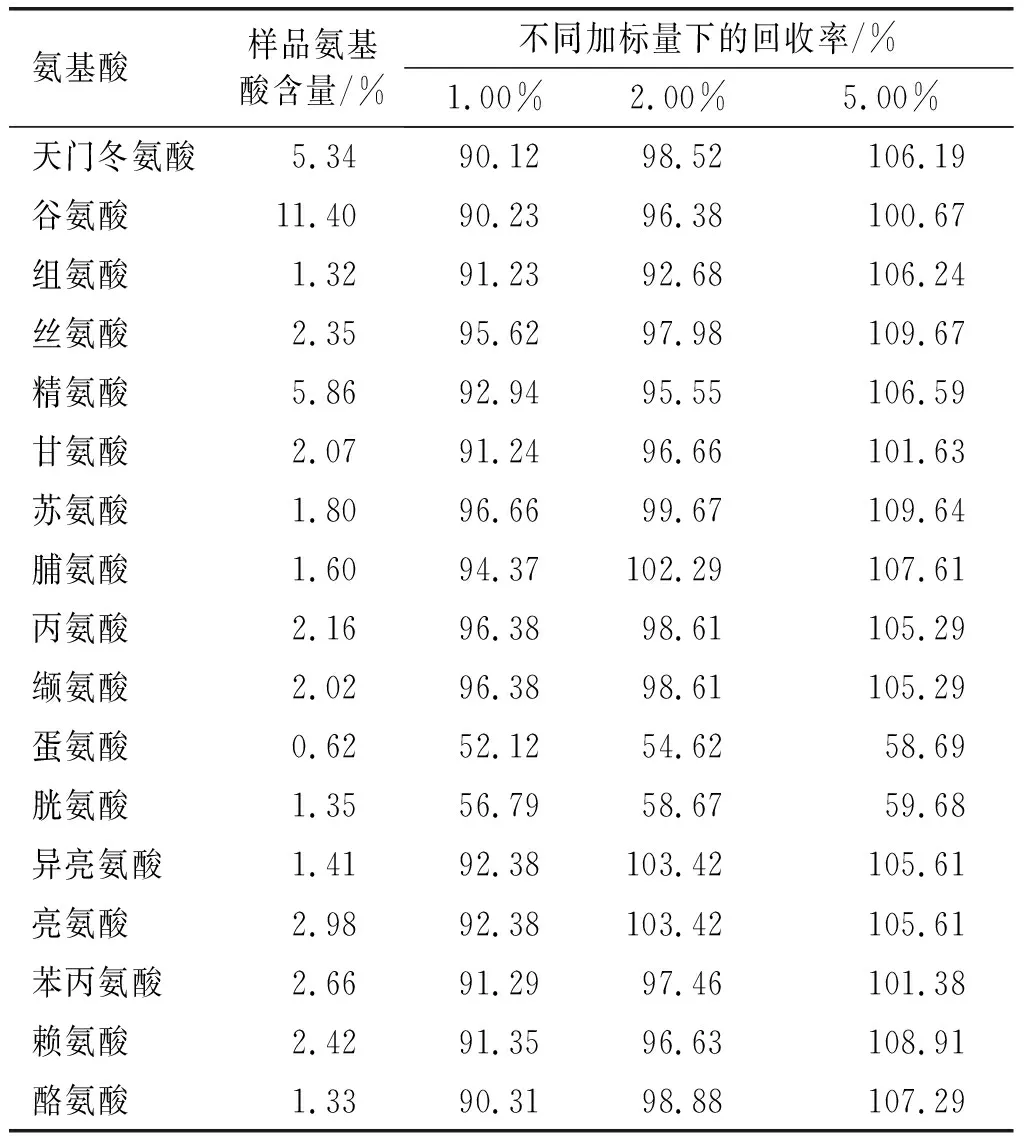
表8 回收率实验结果
由表8可知,蛋氨酸、胱氨酸加标回收率较低,在50%~60%之间,原因同精密度。其余15种氨基酸加标回收率在90.12%~109.67%之间。说明方法除蛋氨酸和胱氨酸外具有较好的准确度。
2.7 实际样品检测
取4批棉籽蛋白样品,按建立的实验方法测定样品中17种氨基酸含量,结果见表9。由表9可知,该方法可以实现棉籽蛋白中17种氨基酸含量的同时检测。
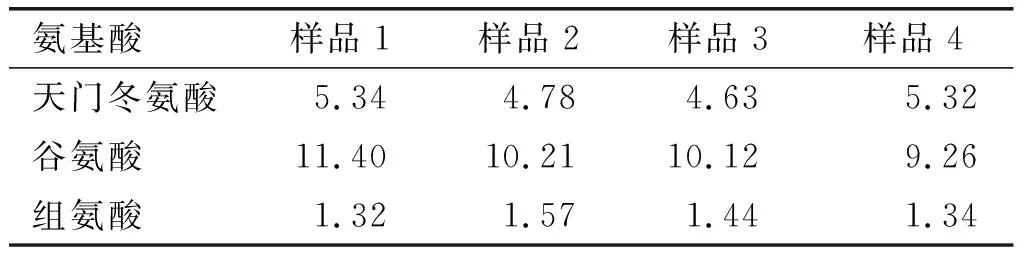
表9 实际样品检测结果 %
3 结 论
通过优化和对比实验,探讨烘箱法酸解样品和微波消解法酸解样品检测结果的准确性,以及微波消解法酸解样品的精密度,优化衍生参数条件、色谱条件,考察了方法的线性范围、检出限、定量限、准确度和精密度等指标,实现了棉籽蛋白中17种氨基酸含量的同时检测。实验结果表明:17种氨基酸在0.004 6~0.045 7 mg/mL范围内呈良好线性关系,相关系数(R2)为0.983 2~0.999 8;精密度除蛋氨酸和胱氨酸较差外,其余15种氨基酸在1.71%~8.43%之间;除蛋氨酸和胱氨酸外,其余15种氨基酸的加标回收率为90.12%~109.67%。本方法操作简单,准确可靠,所用衍生试剂价格便宜、容易获得、过量无干扰,衍生副产物能够与分析物很好地分离,适合于棉籽蛋白中17种氨基酸含量的检测。
猜你喜欢
——全棉籽的加工与利用
