FIRST MOLECULAR EVIDENCE OF CERATOMYXA EPINEPHELA (MYXOZOA:CERATOMYXIDAE) AND ITS GENETIC VARIATION FROM DIFFERENT HOST SPECIES
2020-12-10HUANGYanMeiZHAOYuanJunZHOUYangandYANGChengZhong
HUANG Yan-Mei, ZHAO Yuan-Jun, ZHOU Yang and YANG Cheng-Zhong
(Chongqing Key Laboratory of Animal Biology, Chongqing Normal University, Chongqing 401331, China)
Abstract: Ceratomyxa epinephela Wu, Wu et Hua, 1993 is originally found to infect the gallbladder of Epinephelus akaara Temminck & Schlegel, 1842 from South China Sea, and no molecular data have been provided up to date. In the present study, we detected this species infecting the gallbladder of E. awoara Temminck & Schlegel, 1842 and E. bruneus Bloch, 1793 from East China Sea. For the first time, we offered the SSU rDNA and ITS1 rDNA information for this species and redescribed such species based on the combination of morphological and molecular data. Mature spores of C. epinephela were (4.8±0.5) μm (3.6—5.6 μm)in length and (31.8±4.8) μm (23.3—37.5 μm) in thickness. Myxospores valves were smooth and almost equal, which were joined by a prominent suture. Polar capsules measured (2.9±0.2) μm (2.4—3.7 μm) in length and (2.6±0.2) μm (2.2—3.1 μm) in width. Posterior angle was slightly concave to flat, measured(175.9±3.7)° (165.5°—179.7°). Phylogenetic analysis based on SSU rDNA revealed that C. epinephela had the close relationship with C. nolani Gunter & Adlard, 2009, C. cutmorei Gunter & Adlard, 2009, and C.yokoyamai Gunter & Adlard, 2009, all of whose host were epinephelids. It also implied that the close host affinity might have close phylogenetic relationships of their myxosporean parasites. The sequences analysis based on SSU rDNA and ITS1 rDNA revealed that the four isolates of C. epinephela had already formed different populations, while the species did not form a specialized genetic characteristic among its different host species.
Key words: Ceratomyxa epinephela; Redescription; SSU rDNA; ITS1 rDNA; Genetic variation
Myxosporea is a group of parasites predominantly infecting marine and freshwater fishes. GenusCeratomyxaThélohan, 1892 is one of the largest genera in myxosporea[1,2], with more than 270 species described[3—5].Ceratomyxaspecies possess elongated crescent-shaped or arcuate spores with shell valves often conical, exceeding in length the axial diameter of the spore. Sub-spherical polar capsules have capsular foramina near the suture line at the anterior pole of the spore. Exceptionally, the polar capsules open at opposite sides of the central suture line[6].
About two decades ago, most myxosporeans only have morphological data, making it difficult to differentiate similar species solely based on their morphology since the boundaries of species sometimes are vague, and the morphology of species is always plastic. Therefore, more stable evidence is required for species identification. For nearly two decades, the sequence of small subunit ribosomal RNA (SSU rDNA) has been demonstrated as an effective molecu-lar marker for species identification of Myxozoa[7].Morphology in combination with molecular evidence gives a full consideration of the species independence and also revises many misidentified species, which are diagnosed only by morphological information[8—11].Nowadays, almost of all studies about taxonomy of Myxozoa are conducted by, and recommended, the combination of molecular and morphological data[12].
Epinephelids are important marine economic fishes which widely distributed in tropical, subtropical continental coasts and reefs around the coastal areas[13]. To date, eight species ofCeratomyxahave been recorded to infect epinephelids. They areCeratomyxa cutmoreiGunter & Adlard, 2009,C. hamourMansour, Al-Qahtani, Al-Quraishy & Abdel Baki, 2015,C. hooperiGunter & Adlard, 2009,C. nolaniGunter& Adlard, 2008,C. yokoyamaiGunter & Adlard,2009,C. reniformaWu, Wu et Hua, 1993,C. epinephelaWu, Wu et Hua, 1993,C. guishanensisWu,Wu et Hua, 1993[14—16]. Among them,Ceratomyxa epinephelais originally found to infect the gallbladder ofEpinephelus akaaraTemminck & Schlegel,1842 from South China Sea. This species is only described using its morphological and morphometric data, and no molecular data is available up to date. In the present study, we obtainedC. epinephelafrom the gallbladder ofE. awoaraTemminck & Schlegel 1842 andE. bruneusBloch, 1793 in the coastal waters of Xiamen, East China Sea and sequenced its SSU rDNA and ITS1 rDNA. Here, we redescribed this species by using the combination of morphological and molecular information. Additionally, we analyzed the population variation ofC.epinephelafrom different hosts based on SSU rDNA and ITS1 rDNA data.
1 Materials and methods
1.1 Sample collection and morphological analysis
A total of 11 wildE. awoaraand 19 wildE.bruneuswere collected from the coastal waters of Xiamen, East China Sea, China in July, 2018.Ceratomyxa epinephelawas observed from gallbladder of four host fishes, isolate 1 and isolate 2 were obtained fromE. awoara, and isolate 3 and isolate 4 were obtained fromE.bruneus.Fresh myxospores were rinsed three times with sterile distilled water,followed by centrifugation at 2000 g. Species identification and specimen treatment were carried out according to a previously described method[17]. The specimens were observed, measured and photographed using a Leica DM6000B microscope (Leica Microsystems, CMS GmbH, Germany) at 1000 × magnification. The illustrations, based on fresh materials,were drawn with the aid of a camera lucida and computer software. Measurements, based on 62 spores,were given in microns (μm) and expressed as the arithmetic mean and standard deviation, followed by the range in parentheses. Non-parametric Mann-Whitney test was used to assess the differences of morphological variables between isolates ofC. epinephela.
1.2 DNA isolation, polymerase chain reaction(PCR), cloning and sequencing
Genomic DNA ofC. epinephelawas extracted using the DNeasy Blood and Tissue Kit (QIAGEN,Hilden, Germany) following the manufacturer’s instructions. A portion of the SSU rDNA gene was amplified by PCR in a 20 μL reaction system consisting of 0.7 μL of each primer pair ERIB1 (5′-ACCTG GTTGATCCTGCCAG-3′) and ERIB10 (5′-CTT CCGCAGGTTCACCTACGG-3′)[18], 4.6 μL genomic DNA, 8 μL ddH2O and 6 μL master Mix (Novoprotein, China). Briefly, after an initial denaturation step at 94℃ for 5min, amplifications were carried out with 35 cycles at a melting temperature of 94℃ for 20s, an annealing temperature of 55℃ for 20s and an extension temperature of 72℃ for 2min, followed by a final extension at 72℃ for 5min. ITS1 rDNA was amplified by PCR with primers ERIB10-V (5′-CC GTAGGTGAACG-3′) and 28S1R (5′-GT GTTTCAAGA CGGGTGG-3′)[19]using the above-mentioned PCR procedure. The amplicons were purified using the DNA Agarose Gel Extraction Kit (Omega Bio-Tek,Norcross City, GA). The purified amplicons were inserted into a pMD19-T vector (TaKaRa, Otsu, Japan),and two clones of each amplicon were sent to Invitrogen (Shanghai China) for sequence determination.
1.3 Molecular and phylogenetic analyses
A total of 45 myxosporean SSU rDNA sequences were used in the present phylogenetic analysis, including our four sequences and their highly similar sequences, which were obtained based on Gen-Bank Basic Local Alignment Search Tool (BLAST)search.Tetracapsuloides bryosalmonae(accession No. KF731712) andBuddenbrockia plumatellae(accession No. AY074915) were chosen as outgroups in the phylogenetic tree of SSU rDNA. Multiple alignments were performed by using Clustal X[20]with the default settings. Subsequently, Gblocks[21]was used to remove highly variable, potentially poorly aligned or fast-evolving positions. Bayesian phylogenetic analysis was conducted using MrBayes 3.12[22]. The bestfitting model (GTR + I + G) of sequence evolution for Bayesian analysis was obtained by Modeltest 3.7[23]under the Akaike information criterion (AIC). Four independent Markov chain Monte Carlo (MCMC)analyses were simultaneously run for 3 million generations, sampling one tree per 200 generations, and the first 25% of the samples were discarded as the burnin. At program termination, the average SD of split frequencies was 0.004170. Tracer v. 1.3[24]was employed to check chain convergence and parameter mixing. Maximum likelihood (ML) analysis was conducted using the RAx ML HPC software[25]with 1,000 bootstrap replicates with the GTR+G model.The ITS1 tree ofC. epinephelawas also reconstructed using the same method.
2 Results
Ceratomyxa epinephela(Myxozoa: Ceratomyxidae)
Host (s):E. awoara(Family: Serranidae) andE.bruneus(Family: Serranidae)
Locality: Coastal waters of Xiamen (24°24′56′′N,118°5′51′′E), East China Sea, China
Infection site: Gallbladder, no infection was found in other tissues and organs.
Prevalence: 18.2% (2 of 11) forE. awoara,and 10.5% (2 of 19) forE. bruneus.And the infection intensity for all strains were mild (+) (Tab. 1)
Spore description: mature spores isolated fromE. awoaraandE. bruneuswere elongated and slightly tapering with narrow pointed ends, measured (4.8±0.5) μm (3.6—5.6 μm) in length and (31.8±4.8) μm(23.3—37.5 μm) in thickness (n=62). Spore valves were smooth and almost equal, which were joined by a prominent suture (Fig. 1a-d, f; Tab. 2). Polar capsules were in anterior in position, equal in size and sub-spherical in shape, measured (2.9±0.2) μm (2.4—3.7 μm) in length and (2.6±0.2) μm (2.2—3.1 μm)in width (n=124). Posterior angle was slightly concave to flat, measured (175.9±3.7)° (165.5°—179.7°,n=62) (Fig. 1a-d, f; Tab. 2). Abnormal spores with two short shell valves were also observed (Fig. 1e).Morphometry comparison showed that there were nosignificant differences (P>0.05) in spore length, spore thickness, polar capsule length, polar capsule width and posterior angle ofC. epinephelafromE. awoaraandE. bruneus.
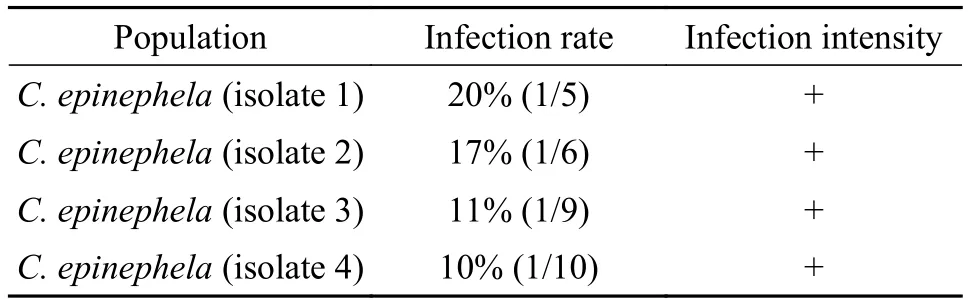
Tab. 1 Infection data of the C. epinephela from E. awoara and E.bruneus
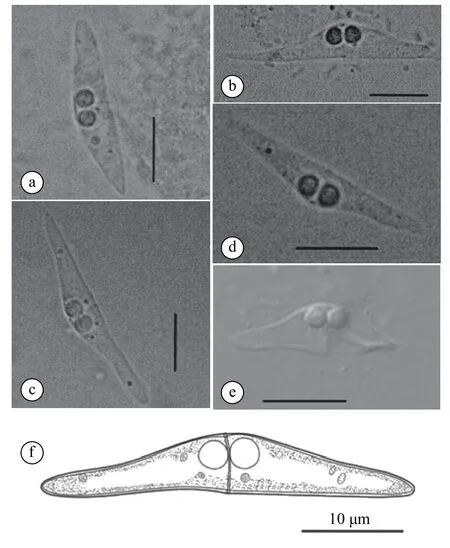
Fig. 1 Spores of C. epinephela: a and b show the mature spores from E. awoara; c and d show mature spores from E. bruneus; e shows abnormal spore from E. bruneus; f is the line drawings of mature spore
Molecular characterization: the partial SSU rDNA sequences of four isolates ofC. epinephelawere 1575 nt (isolate 1), 1575 nt (isolate 2), 1615 nt(isolate 3) and 1564 nt (isolate 4) in length, and their GenBank access numbers were MN541835, MN 541836, MN541837 and MN541838, respectively.GenBank BLASTn search revealed that SSU rDNA sequences of the four isolates ofC.epinephelawere unique among all myxozoans. The species with the highest similarity (96.5%—96.8%) and the lowest genetic divergence (0.027—0.029) toC. epinephelawasC. nolaniEU728698, followed byC. yokoyamaiEU729696 (95.5%—96.6%, 0.034—0.036) (Tab. 3).The SSU rDNA similarity and genetic divergence of the four isolates were 99.7%—99.9% and 0.001—0.003, respectively (Tab. 3).
The partial ITS1 rDNA sequences of the four isolates ofC.epinephelawere 728 nt (isolate 1), 755 nt(isolate 2), 755 nt (isolate 3) and 755 nt (isolate 4) in length, and their GenBank access numbers were MN541824, MN541825, MN541826 and MN541827,respectively. The ITS1 rDNA similarity and genetic divergence of the four isolates were 99.0%—99.9%and 0.001—0.010, respectively (Tab. 4). The four samples had eight variation sites (12, 17, 33, 408,424, 431, 494 and 510 site), indicating that these samples belonged to different genotypes and belonged to different populations.
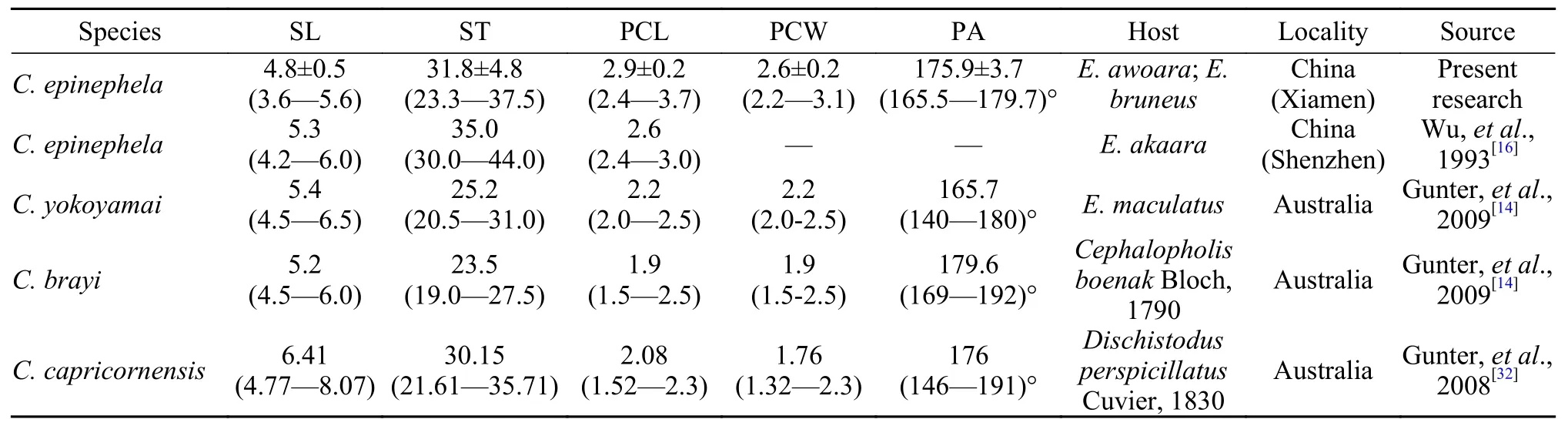
Tab. 2 Comparison of C. epinephela with the similar Ceratomyxa species in morphology
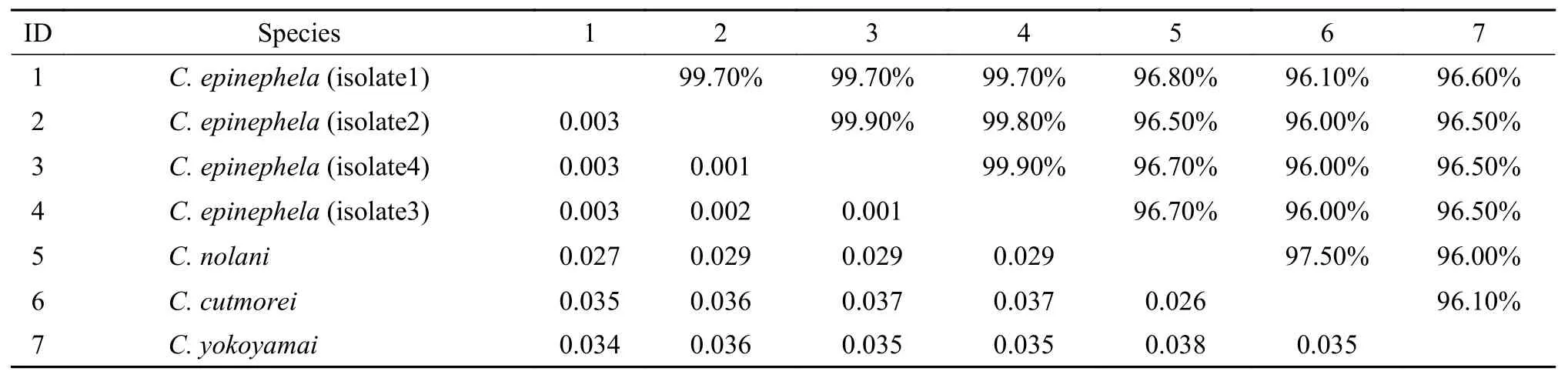
Tab. 3 Genetic divergence (lower left triangle) and sequence similarities (upper right triangle) of C. epinephela with the similar Ceratomyxa species based on SSU rDNA sequences

Tab. 4 Genetic divergence (lower left triangle) and sequences similarities (upper right triangle) of C. epinephela based on ITS1 rDNA sequences
Phylogenetic analysis: the phylogenetic tree based on SSU rDNA sequences inferred from the BI and ML methods exhibited the same topology (Fig. 2A).In the phylogenetic tree, the four isolates ofC. epinephelawere clustered as a clade, which nested in the assemblage of epinephelid ceratomyxids (composed ofC. nolaniEU728698,C. yokoyamaiEU729696 andC. cutmoreiEU729691) (Fig. 2A). Within this assemblage,C. yokoyamaiEU728698 diverged earlier,and thenC. cutmoreiEU729691,C. nolaniEU728698 andC. epinephelawere successively separated (Fig.2A). The phylogenetic tree ofC. epinephelabased on ITS1 rDNA sequences revealed that isolate 1 and isolate 3 were clustered together as a clade, which was sistered to the clade consisting of isolate 2 and isolate 4 (Fig. 2B).
3 Discussion
Myxospores ofC. epinephelasampled from Xiamen in this study (hereafter called Xiamen samples)morphologically resembled samples from Shenzhen(hereafter called Shenzhen samples)[16],C. yokoyamai,C. brayiGunter & Adlard, 2009,C. capricornensisGunter & Adlard, 2008,C. auerbachiKabata, 1962.Overall, the Xiamen samples were most morphologically consistent with Shenzhen samples, although the Xiamen samples (thickness: 23.3—37.5 μm) were slightly thinner than Shenzhen samples (thickness:30.0—44.0 μm) (Tab. 1).C. epinephelacould be distinguished fromC. capricornensisby its shorter length[(3.6—5.6) μmvs. (4.77—8.07) μm]and larger polar capsules [(2.4—3.7) μm × (2.2—3.1) μmvs. (1.52—2.3) μm × (1.32—2.3) μm, Tab. 1]. The spore sizes ofC. epinephela[(3.6—5.6) μm × (23.3—37.5) μm]were larger than those ofC. yokoyamai[(4.5—6.5) μm ×(20.5—31.0) μm]andC. brayi[(4.5—6.0) μm × (19.0—27.5) μm)]. Xiamen samples ofC. epinephelawere found to infectE. awoaraandE. bruneus, which were different from the Shenzhen samples infectingE.akaaradescribed by Wu,et al., 1993. All host fish ofC. epinephelabelonged toEpinephelus, and the loca-lities of the two geographic samples (Xiamen samples and Shenzhen samples) were not far away from each other (500 kilometers around). Considering the morphology, morphometry, coupled with host affinity and geographical localities, the present isolates were regarded asC. epinephela.
Phylogenetic analysis based on SSU rDNA showed that the basal divergence within the ceratomyxids wasCeratonovaspecies (Branch a) and the remaining species (Branch b and Branch c). Branch a was composed ofCeratonova shastaAtkinson, Foott &Bartholomew, 2014 andCeratonova gasterostea, Atkinson, Foott & Bartholomew, 2014 which are freshwater species infecting the intestine of their hosts.Branch b consisting ofUnicapsulocaudum mugilumYang, Zhou, Zhao, Huang & Huang, 2017,C.amazonensisMattews, Naldoni, Maia & Adriano,2016,C. gracillimaZatti, Atkinson, Maia, Bartholomew & Adriano, 2018,C. tunisiensisThabet, Mansour, Omar & Tlig-Zouari, 2016,C. leatherjacketiFiala, ková, Kodádková, Freeman, Bartošová-Sojková et Atkinson, 2015, andMyxodavisia bulaniFiala, Hlavnicková, Kodádková, Freeman, Bartosová-Sojková & Atkinson, 2015 represented a transition lineage[2].Ceratomyxa epinephelawas clustered in a more recent divergent lineage (Branch c), which possessed a majority of typical gallbladder-infecting marine ceratomyxids. Within this lineage,C. epinephelanested in an assemblage formed byC. nolani,C. cutmoreiandC. yokoyamai, the hosts of which were epinephelids. It seemed that close host affinity might have the close phylogenetic relationships of their parasites. As a matter of fact, many previous studies have also indicated that the host affinity may play a crucial role in the evolution of myxosporean species,and the close host affinity may have close relationship of myxosporean species[5,26,27]. However,C.hooperiGunter & Adlard, 2009 andC. hamourMansour, Al-Qahtani, Al-Quraishy & Abdel-Baki, 2015,the hosts of which are species ofEpinephelus, were not clustered together in the main assemblage of epinephelid ceratomyxids stated above. We speculated thatC. hooperiandC. hamourmight have migrated from other fish host (rather than epinephelids) in their evolution history. In other words, they might have experienced host shift.
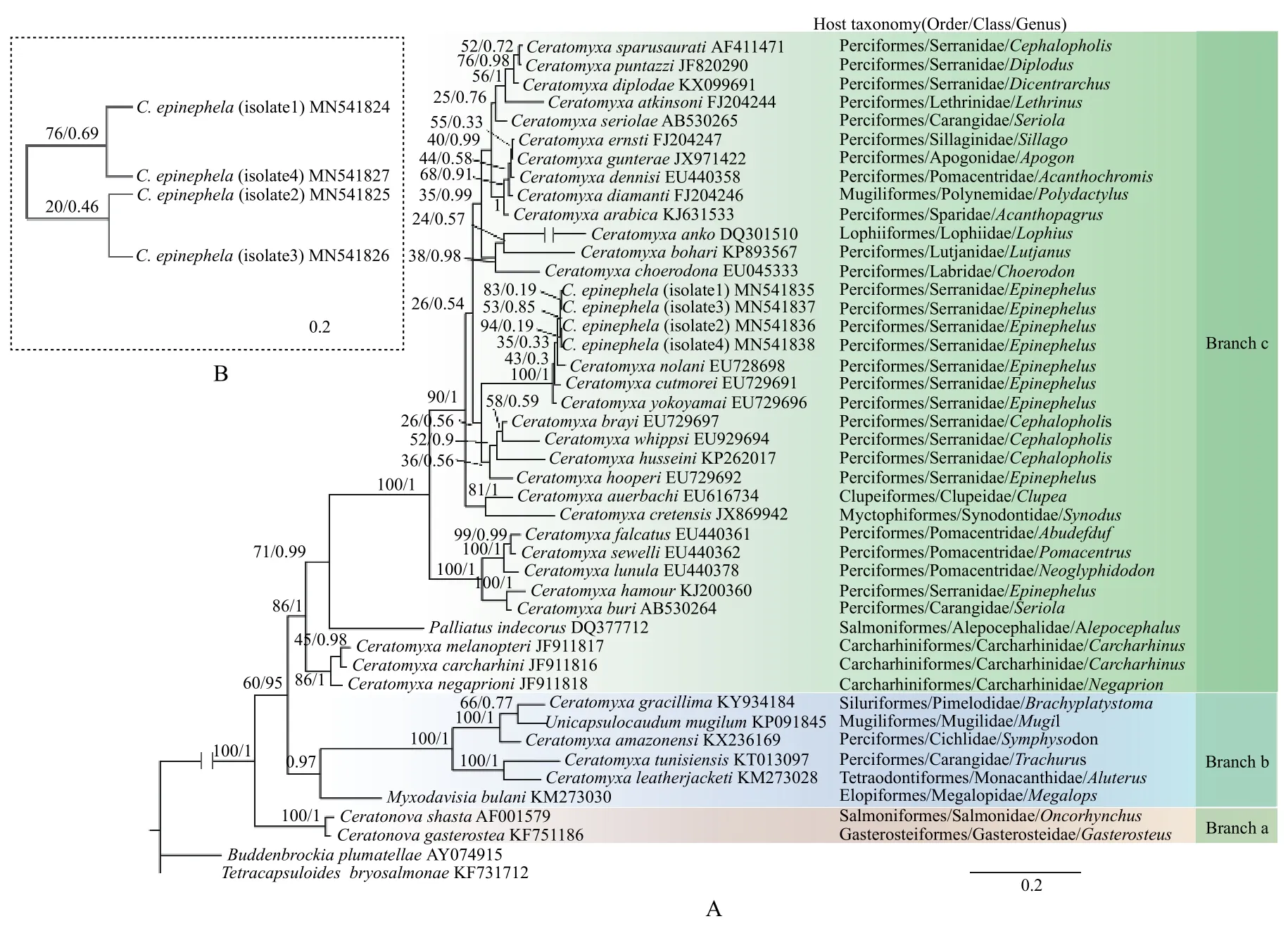
Fig. 2 Molecular phylogenetic trees constructed using maximum likelihood (ML) analysis and Bayesian inference (BI)
Generally, geographic isolation plays an important role in population divergence for free-living animals, and the longer geographic distance commonly causes stronger divergence between populations.However, the population divergence is not only related to geographic isolation, but also associated with host species, infection sites, inhabits and so on for parasites[28]. Many previous studies have implied that some parasites may be specialized among their different host species, and stronger genetic differentiation has been found between populations with different host species compared with that between geographically isolated populations in the same host species[28—31].However, phylogenetic analysis based on SSU rDNA and ITS1 rDNA data showed that the isolate 1 and isolate 2 collected fromE. awoarawere not clustered together, and the same situation was found for the isolate 3 and isolate 4, which were collected fromE.bruneus(Fig. 2A, B). The results from analyses of variation sites, genetic divergence and sequence similarity based on ITS1 rDNA also showed that there was no special genetic relationship for the isolates from the same host species. These results implied thatC. epinepheladid not formed a specialized genetic characteristic among its different host species. The reasons might be following two (1) the populations ofC. epinephelainfectE. awoaradid not infectE.bruneus, and vice versa, and the time was not long enough forC. epinephelapopulations from different host species to form a distinct genetic characteristic or(2) the individuals ofC. epinephelamight continuously and mutually migrate betweenE. awoaraandE. bruneus, as the two host species coexisted in the same region. Therefore, the genetic differentiation ofC. epinephelafromE. awoaraandE. bruneusmight be weakened.
