基于GEO芯片联合网络药理学探讨当归饮子治疗慢性荨麻疹的cross-talk及miRNA-mRNA机制*
2020-12-08包成通魏歆然张依依匡泓俊牛子青刘小娟李俊熙
包成通,魏歆然,张依依,钟 峰,匡泓俊,牛子青,刘小娟,王 璐,李俊熙,章 薇△
(1.湖南中医药大学,湖南 长沙 410208;2.贵州中医药大学,贵州 贵阳 550025;3.湖南中医药大学第一附属医院,湖南 长沙 410007)
慢性荨麻疹(CU)[1]是因皮肤黏膜的小血管短暂通透性增加,而出现局限性水肿的一种常见慢性疾病,以易反复发作,迁延不愈,难以根治为主要特征,以皮肤风团、瘙痒、水肿等为主要临床表现,发病机制主要与自身免疫、感染、炎症反应等相关。CU属中医学“白疹”“瘾疹”“风疹”等范畴[2],病机总属本虚标实,与血虚风燥相关,气血亏虚,卫外失护,开泄失度,外邪(风邪为主)乃入;气血阻滞,精液不畅,布达难荣,化燥生风,风盛则瘙痒,正邪交争于皮肤腠理,发为瘾疹[3]。当归饮子出自《重订严氏济生方》[4],由荆芥、黄芪、白蒺藜、何首乌、防风合四物汤组成,具有养血和营、祛风止痒之功,皮肤疥疮,或痒或肿,或脓或发赤者适用,临床上治疗CU具有较好疗效,主要与调节免疫、抑制炎症反应有关,但深层机制尚不明确。本研究基于生物信息学技术,采用GEO芯片联合网络药理学的方法,初步探讨当归饮子治疗CU的cross-talk及miRNA-mRNA深层机制,为后期实验及临床验证提出理论现实依据,为CU的临床治疗提供潜在靶点。
1 材料与方法
1.1 慢性荨麻疹差异基因分析 以慢性荨麻疹“Chronic Urticaria”为检索词,在NCBI基因表达GEO数据库(https://www.ncbi.nlm.nih.gov/geo/)中检索,获取GSE57178芯片表达数据集,该数据集中包括6个病变皮肤样本,7个非病变皮肤样本,5个正常皮肤样本。本研究采用R语言“limma”包对比分析6个病变皮肤样本(GSM1376737、GSM1376739、GSM1376741、GSM1376743、GSM1376745、GSM1376747) 与 5个正常皮肤样本 (GSM1376750、GSM1376751、GSM 1376752、GSM1376753、GSM1376754),以│logFC│>1;P-value<0.05为筛选条件,筛选出慢性荨麻疹的差异基因(Differential Expression Genes;DEGs),并绘制DEGs火山图。
1.2 当归饮子治疗慢性荨麻疹潜在靶点筛选 通过TCMSP[5(]http://lsp.nwu.edu.cn/tcmsp.php)、TCMID(http://www.megabionet.org/tcmid/)数据库,以口服生物利用度(OB)≥30%;药物类药性(DL)≥0.18为筛选条件[6-7],获取当归饮子中药有效成分。
通过PubChem[8(]https://pubchem.ncbi.nlm.nih.gov/)数据库收集当归饮子中药有效成分SDF结构式。
通过Swiss Target Prediction[9(]http://www.swis stargetprediction.ch/)数据库,将当归饮子中药有效成分SDF结构式导入,获取当归饮子潜在靶点。
通过R语言“VennDiagram”包绘制当归饮子潜在靶点与慢性荨麻疹DEGs韦恩图,以获取当归饮子治疗慢性荨麻疹的潜在靶点。
1.3 当归饮子治疗慢性荨麻疹潜在靶点PPI网络构建及拓扑分析 通过String[10(]https://string-db.org/)数据库,导入当归饮子治疗慢性荨麻疹的潜在靶点,获取PPI网络相关信息,采用Cytoscape 3.7.2软件对PPI信息进行网络可视化。采用“Network Analysis”对PPI分析,设定 0<Degree≤5 为“粉红色”;5<Degree≤10 为“蓝色”;10<Degree≤15“大红色”;“Degree”值越大则节点越大,可信分数越高则线条越粗;“Combined Score”值越大,边越粗。采用“CytoHubba”对 PPI进行“Closeness”、“Betweenness”、“MCC”拓扑分析,以筛选出当归饮子治疗慢性荨麻疹的关键靶点(Hub-gene)。
1.4 当归饮子治疗慢性荨麻疹的cross-talk机制初探通过DAVID[11](https://david.ncifcrf.gov/summary.jsp)数据库,导入当归饮子治疗慢性荨麻疹的潜在靶点,获取KEGG富集分析数据。采用R语言“ggplot2”包,以富集倍数为横轴;通路为纵轴;-lgP为颜色;基因数为气泡大小绘制KEGG富集分析气泡图。从当归饮子治疗慢性荨麻疹的KEGG通路中,筛选出荨麻疹最相关通路,采用R语言“circlize”包绘制“基因—KEGG”弦图,以寻找出当归饮子治疗慢性荨麻疹关联性最强的通路靶点,同时采用 R语言“UpSetR”包绘制“基因—KEGG”UpSet图,以筛选出当归饮子治疗慢性荨麻疹的具体cross-talk通路靶点。
1.5 当归饮子治疗慢性荨麻疹的miRNA-mRNA机制初探利用TargetScan[12](http://www.targetscan.org/mamm_31/)数据库反向预测当归饮子治疗慢性荨麻疹潜在靶点mRNA的miRNA,筛选并收集保守性miRNA,同时利用Cytoscape 3.7.2软件构建miRNA-mRNA相互作用网络调控模式。设定miRNA为方块、灰色;mRNA为菱形、红色。
2 结果
2.1 慢性荨麻疹差异基因分析结果 采用R语言limma包对GEO芯片进行差异分析,以│logFC│>1;P-value<0.05为筛选条件获取DEGs,分析出257个差异基因,其中240个基因表达上调(如:HIF1A、MMP12、STAT3等);有17个基因表达下调(如:FABP7、KRT15、LGR5 等)。以“logFC”为横轴,“-lgP”为纵轴,差异上调基因设为“红色”,差异下调基因设为“蓝色”,绘制 DEGs火山图(图 1)。

图1 慢性荨麻疹差异基因火山图
2.2 当归饮子治疗慢性荨麻疹潜在靶点筛选结果 通过TCMSP、TCMID数据库,共获取当归饮子中药有效成分共202个,其中黄芪15个(如:Mairin、Jaranol、Bifendate等),白芍 13个(如:Paeoniflorgenone、Lactiflorin、beta-sitosterol等),川芎 7个(如:Wallichilide、Perlolyrine、Myricanone等),当归 2 个(如:beta-sitosterol、Stigmasterol),防风 18 个(如:Anomalin、Divaricatacid、Ammidin等),何首乌 73个(如:beta-sitosterol、Tricin、Rhein等),蒺藜 12个(如:Terrestriamide、Isorhamnetin、Sitosterol等),荆芥 11个(如:Diosmetin、Sitosterol、Schizonepetoside B 等),生地黄 51个(如:Acteoside、Daucosterol、Glutinoside等)。
通过PubChem、Swiss Target Prediction数据库,对上述202个当归饮子中药有效成分进行靶点预测,去除重复值,保留唯一值后,共获取964个当归饮子潜在靶点。采用R语言“VennDiagram”包绘制当归饮子潜在靶点与慢性荨麻疹DEGs韦恩图(图2),获取36个当归饮子治疗慢性荨麻疹的潜在靶点(表1)。
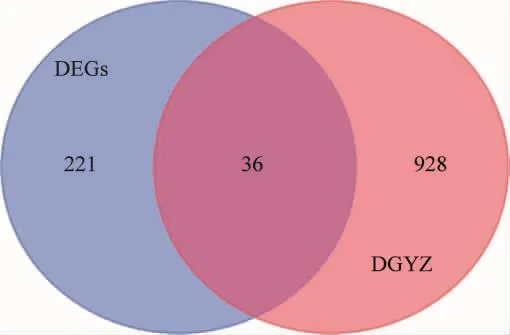
图2 当归饮子潜在靶点与慢性荨麻疹DEGs韦恩图
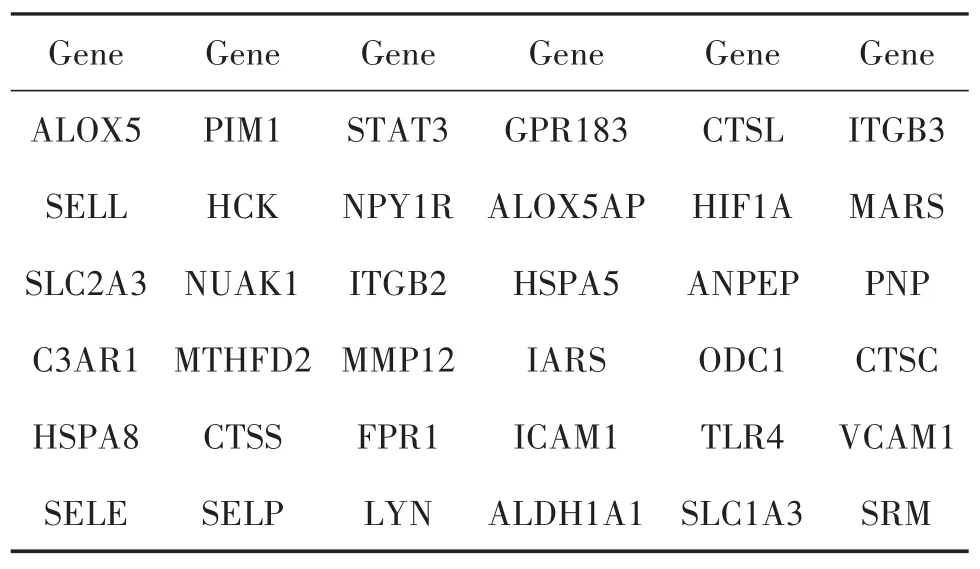
表1 当归饮子治疗慢性荨麻疹的潜在靶点
2.3 当归饮子治疗慢性荨麻疹潜在靶点PPI网络构建及拓扑分析结果 通过String数据库,采用Cytoscape 3.7.2软件对PPI信息进行网络可视化及网络分析(图3),该PPI网络中,共有36个节点,91条边,P<1.0e-16,平均节点度为5.06,平均聚类系数为0.584。根据 Degree 值排序显示:ITGB2>TLR4>STAT3>VCAM1>10,表示此4个靶点在当归饮子治疗慢性荨麻疹中作用最强。同时采用“CytoHubba”对PPI进行“Closeness”、“Betweenness”、“MCC”拓扑分析(图 4),以筛选出 Hub-gene(表 2)。
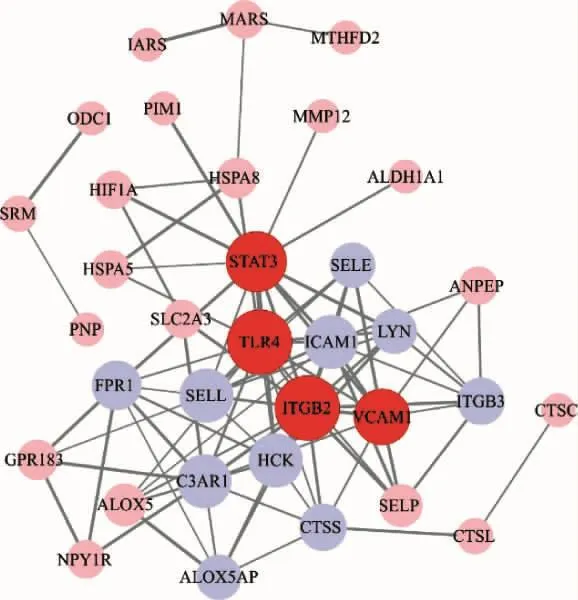
图3 当归饮子治疗慢性荨麻疹潜在靶点PPI网络图

表2 当归饮子治疗慢性荨麻疹Hub-gene
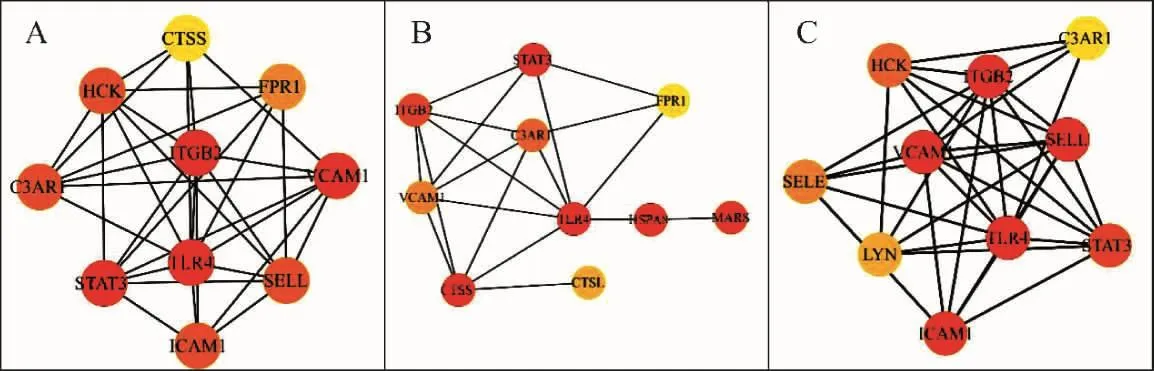
图4 当归饮子治疗慢性荨麻疹潜在靶点拓扑分析网络图
2.4 当归饮子治疗慢性荨麻疹的cross-talk机制 通过DAVID数据库,获取KEGG富集分析数据,采用R 语言“ggplot2”“circlize”“UpSetR”包,分别绘制“基因—KEGG”气泡图、弦图、UpSet图。当归饮子治疗慢性荨麻疹潜在靶点共富集17条KEGG通路,如气泡图所示(图5A):NF-kappa B通路、HIF-1通路、TNF通路等。通路富集的基因靶点,如弦图所示 (图 5B):NF-kappa B 通路由 LYN、VCAM1、TLR4、ICAM1基因靶点富集;HIF-1通路由STAT3、HIF1A、TLR4基因靶点富集;TNF通路由VCAM1、SELE、ICAM1基因靶点富集等。cross-talk是通路共同基因靶点相互“串话”,激活2条以上通路,介导一种或多种表型的机制模式,当归饮子治疗慢性荨麻疹潜在靶点中cross-talk机制如UpSet图所示(图5C):1个靶点(SELE)在TNF通路与细胞粘附分子(CAMs)2条通路中“串话”;1个靶点(ITGB2)在吞噬体与CAMs 2条通路中“串话”;2个靶点(CTSL、CTSS)在抗原处理表达、吞噬体与溶酶体3条通路中“串话”;2个靶点(VCAM1、ICAM1)在 NF-kappa B、TNF与CAMs 3条通路中“串话”;1个靶点(TLR4)在 HIF-1、NF-kappa B、吞噬体 3条通路中“串话”。
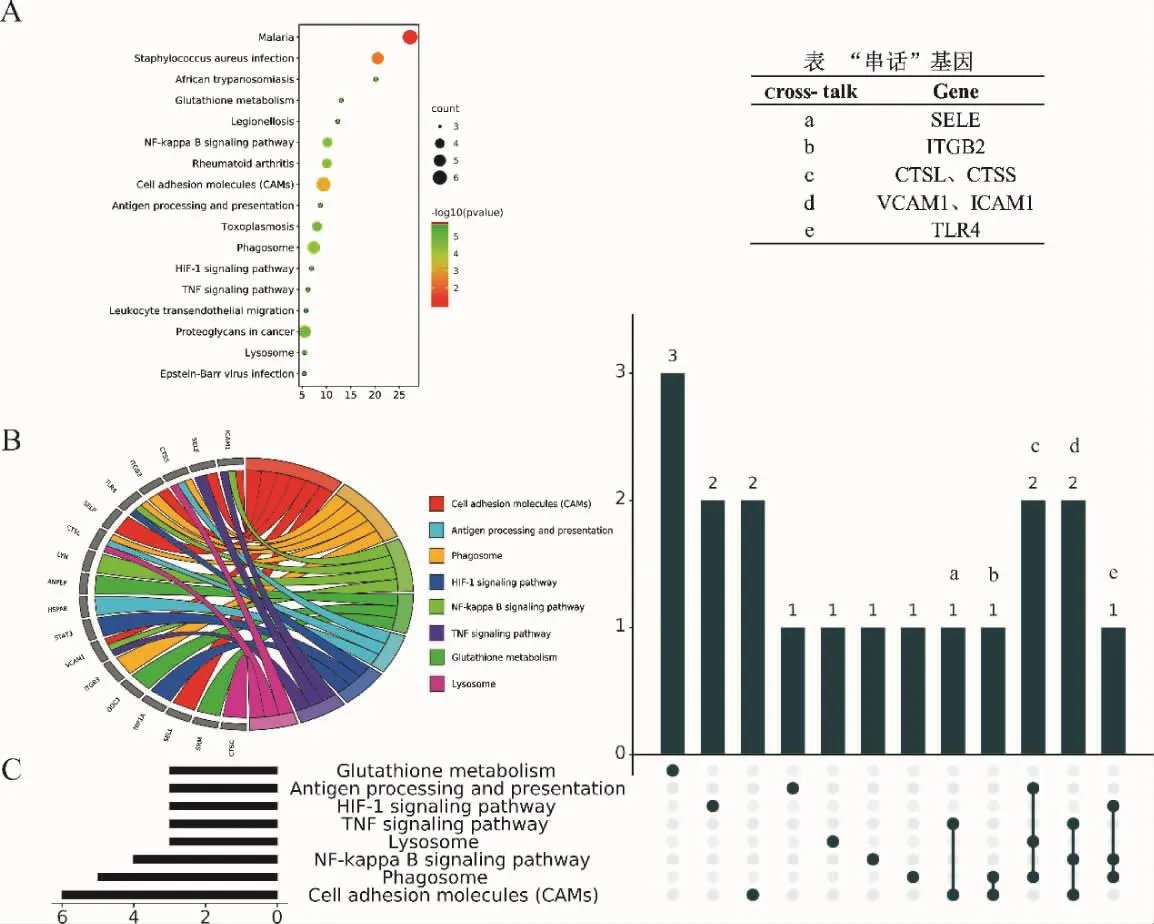
图5 当归饮子治疗慢性荨麻疹“基因—KEGG”气泡图(A)、弦图(B)、UpSet图(C)
2.5 当归饮子治疗慢性荨麻疹的miRNA-mRNA机制 TargetScan数据库反向预测当归饮子治疗慢性荨麻疹的miRNA,其中36个潜在靶点中,共有24个潜在靶点 mRNA(如:TLR4、HIF1A、STAT3等)存在保守 miRNA(如:miR-140-5p;miR-542-3p;miR-489-3p等)调控,构成了477组miRNA-mRNA模式(如 :miR-140-5p-TLR4;miR-199a-5p-HIF1A;miR-370-3p-STAT3等),相互作用结果如图6。
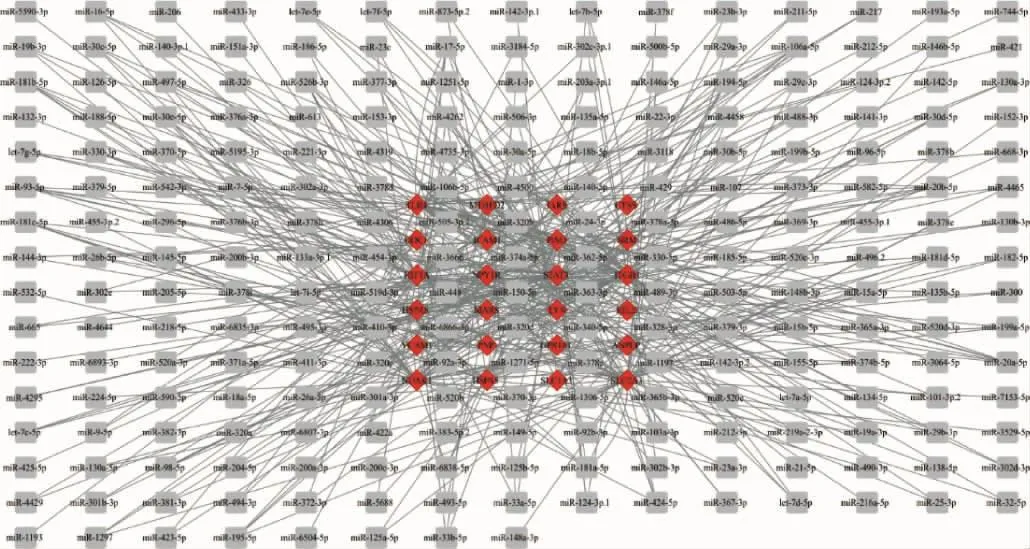
图6 相互作用结果
3 讨论
cross-talk即“串话”[13],原指通信线路上信号杂散耦合到其他通信线路造成干扰的现象,在细胞信号通路相关研究中,认为细胞间信号通路是一个复杂的网络体系,单一信号通路在一定条件下会受到其它信号通路的影响,从而产生了细胞生物学界的“信号串话”。 miRNA[14]是微小的非编码RNA,主要通过结合靶mRNA的3’非翻译区(UTR),导致靶mRNA进行切割或转录抑制。在CU的治疗中cross-talk与miRNA-mRNA机制是当前的热点问题。
当归饮子由荆芥、黄芪、白蒺藜、何首乌、防风、当归、川芎、白芍、生地黄组成,方中四物汤养血活血,补通同用;何首乌养血滋阴,祛风止痒;黄芪益气实表,托毒行滞;白蒺藜、防风、荆芥散风邪以止痒,全方共奏养血、滋阴、润燥、祛风之功,善治CU之血虚风燥证。当归饮子治疗CU血虚风燥证具有较好的疗效,且在实验中得到广泛验证,叶静静[15]对168例CU患者进行RCT,发现当归饮子能显著降低患者血清中MDA含量,抗氧化应激以提高免疫,从而降低CU复发率,改善患者瘙痒、水肿等症状。徐风等[16]研究证实当归饮子能下调LC3II、p62表达,促进自噬小体形成,改善CU水肿及毛细血管扩张,但其深层机制尚未明确,需待进一步研究。
本研究旨在利用生物信息学算法研究当归饮子治疗CU的cross-talk及miRNA-mRNA机制。GEO芯片差异分析出257个差异基因,240个基因表达上调,17 个基因表达下调(如:FABP7、KRT15、LGR5等)。网络药理学方法分析出当归饮子有效成分及潜在靶点共964个,取潜在靶点与差异基因交集后得到36个当归饮子治疗CU的潜在靶点;对筛选分析出的当归饮子治疗 CU的潜在靶点进行 PPI、KEGG、cross-talk及miRNA预测分析,以初步明确当归饮子治疗CU的分子机制。
当归饮子治疗CU的PPI网络分析中,有36个节点(基因),91条边,拓扑分析出 TLR4、STAT3、ITGB2等18个关键基因,能初步认为此18个基因可能在当归饮子治疗CU中作用最强。研究发现CU患者外周血中TLR4显著升高,TLR4能诱导IL-6和MIP-1α分泌,介导CU患者免疫炎症反应[17],同时研究发现当归饮子中有效成分当归多糖[18]在多种疾病中能抑制TLR4表达[19-20],抑制炎症反应。Luo等[21]研究发现CU患者皮肤组织中JAK2/STAT3信号通路被激活,炎症细胞增殖,此过程能被OSMR能被抑制,同时王丽新[22]提出STAT3是检测CU临床疗效的潜在客观指标之一。因此可认为36个当归饮子治疗CU的靶点中的18个可作为潜在的临床疗效标志物及治疗靶点。
cross-talk是通路共同基因靶点相互“串话”,激活2条以上通路,介导一种或多种表型的机制模式,本研究以KEGG富集分析为切入点,结合关联性弦图及UpSet图,将筛选出的“通路串话”靶点进行可视化(如:TLR4可能在HIF-1、NF-kappa B、吞噬体3条通路中“串话”等)。Pei等[23]研究发现IL-38能通过HIF-1与NF-kappa B信号通路,降低HIF-1α、NFΚB p65与TLR4表达,抑制炎症反应,在大鼠模型中初步验证了TLR4可能参与HIF-1与NF-kappa B信号通路之间的串话,在炎症反应中发挥作用。Hu等[24]研究发现内皮细胞表面的双链蛋白聚糖(BGN)与其受体TLR2、TLR4结合,激活HIF-1与NF-kappa B信号通路,增强HIF-1α、NF-κB启动子之间的相互作用,从而刺激下游VEGF表达。本研究构建的cross-talk“通路串话”机制模式,虽已有部分在实验中验证,但仍有大量的cross-talk模式尚未研究,因此不仅可作为当归饮子治疗CU的分子机制,而且也为其他疾病的治疗提供了新的思路。
miRNA是一类广泛存在于动植物中,对转录后基因表达调控起关键作用的内源性小非编码RNA。成熟miRNA先与RNA诱导沉默复合体(RISC)的复合物结合,再特异性与下游靶mRNA的3’UTR形成不完全互补配对,阻断转录后翻译,从而调控基因表达,此过程称为miRNA-mRNA互作模式。本研究初步筛选出TLR4、HIF1A、STAT3在内的24个潜在靶点的477组miRNA-mRNA模式(如:miR-140-5p-TLR4;miR-199a-5p-HIF1A;miR-370-3p-STAT3等)。Ching-Kow等[25]通过miRNA微阵列技术发现CU患者中有16种差异miRNA(DEMs),其中5个miRNA(miR-2355-3p、261-3p、4264、2355-5p、29c-5p)与7个上调mRNA(FBXL20,OPHN1,YPEL2,STARD9,SELE,KLHL24,ING4) 相互作用明显,对CU疾病的诊断与治疗提出了指导性的建议,构建的miRNA-mRNA模式中与本研究筛选的miR-6835-3p-SELE、miR-6893-3p-STAT3;miR-140-5p-TLR4等相互吻合,因此本研究构建的当归饮子治疗CU的miRNA-mRNA模式能成为后续实验验证的潜在分子机制模式。
综上所述,本研究采用GEO芯片联合网络药理学的方法,筛选出当归饮子治疗CU的关键靶点,构建了蛋白相互作用的PPI网络,并进一步从通路、cross-ralk、miRNA-mRNA机制角度得出当归饮子治疗CU可能通过 TLR4参与的HIF-1、NF-kappa B、吞噬体3条通路中“串话”机制发挥作用,同时miR-140-5p-TLR4的相互作用关系可能在其中发挥重要作用,同时得出当归饮子治疗CU的多靶点、多cross-ralk、多miRNA-mRNA机制将成为未来机制研究的新方向。本研究从基因、分子角度预测和分析了当归饮子治疗CU的作用机制,为后续研究提供了依据。
