雄激素受体抑制剂治疗前列腺癌的研究进展
2020-12-08杨娴
杨娴
(石家庄四药有限公司,河北 石家庄)
0 引言
前列腺癌(Prostate cancer,PCa)是男性最常见的癌症之一,据统计,2018年全世界有130万例新癌症病例和359000例死亡病例[1]。到2040年,由于人口的增长和老龄化,全世界PCa患者将增加到230万例,死亡病例预计增加74万例[2]。相较于西方发达国家,中国前列腺癌患者晚期比例较高[3]。
雄激素受体(androgen receptor,AR) 抑制剂类药物是治疗前列腺癌特别是晚期前列腺癌的重要药物[4]。AR抑制剂按结构分为2类:甾体类与非甾体类。甾体类受体抑制剂的代表药物为醋酸环丙孕酮,但其口服生物利用度和选择性都比较差,并易产生交叉反应等不良反应,不利于临床。非甾体类雄激素受体抑制剂(Non-steroidal androgen receptor inhibitors,NSARI)的口服生物利用度较高,且对AR的选择性良好,是近年来AR抑制剂类抗前列腺癌药物的主要研究方向。因此,本文对靶向AR受体的非甾体小分子药物的研究进展进行综述。
1 雄激素受体(AR)
1.1 AR的结构
前列腺的发育和功能依赖于雄激素(睾酮和DHT)通过AR信号通路的调节。AR是一个110KD的核受体,基因位于X染色体q臂(Xq11-12)上。全长AR(AR-FL)包含四个结构域:N-基末端结构域(NTD)、DNA结合结构域(DBD)、铰链区和配体结合结构域(LBD)[5-6]。NTD由外显子1编码,有助于AR基因的转录调控;DBD由外显子2和3编码,有助于与DNA结合;铰链区有一个核定位序列(NLS),该序列暴露于雄激素结合上。LBD由外显子4-8编码,通过配体结合介导转录激活[7]。
1.2 AR的激活与相关信号通路
在没有配体(雄激素)的情况下,LBD有助于AR在胞质中的定位。在细胞质内,AR与热休克蛋白(HSP)包括HSP90(特别是HSP90伴侣复合物)、HSP70、HSP56和P23结合,保持失活构象[5]。一旦雄激素与AR的LBD结合,就会引起构象变化,导致HSP90复合物解离,形成同源二聚体,暴露NLS,AR从细胞质向细胞核中转移[8]。在细胞核中,AR通过DBD与靶基因启动子或增强子中雄激素反应元件(AREs)结合,招募调节靶基因转录的因子,其中包括前列腺特异性抗原(PSA)、跨膜蛋白酶丝氨酸2 (TMPRSS2)、Myc原癌基因蛋白(Myc)、血管内皮生长因子(VEGF)、白介素-6蛋白激酶Cδ(PRKCD)以及一些参与类固醇代谢和氧化还原调节的基因[7,9]。通过AR影响基因转录,导致许多细胞过程的增强,如血管生成增加,上皮细胞增殖增加和凋亡减少[9]。
近年来的研究表明,在没有配体的情况下,AR可被其他信号转导途径包括MAPK、Src、PKC、PI3K/Akt和Wnt等信号通路激活。蛋白激酶C(PKC)在没有雄激素的情况下可促进AR核移位。非受体酪氨酸激酶(Src)可在AR的铰链区磷酸化AR,增加AR核转运[8]。PI3K/Akt可以诱导FOXO3a向细胞核移位,FOXO3a与AR启动子结合,增强AR基因表达[10]。生长因子和细胞因子介导的细胞通路能够激活配体无关的AR信号。IL-6与IL-6受体结合导致MAPK和STAT3介导的AR转录激活。同样,在没有雄激素的情况下,EGF也通过RAS/RAF/MAPK和PI3K/Akt途径转录激活AR[11]。
2 非甾体雄激素受体抑制剂(Non-steroidal androgen receptor inhibitors,NSARI)
AR受体抑制剂通过竞争性和选择性地与AR结合,阻断内源性雄激素的结合,阻断雄激素依赖性细胞级联反应,抑制前列腺癌细胞生长。
2.1 第一代NSARI
第一代NSARI包括氟他胺(Flutamide)、尼鲁他胺(Nilutamide)和比卡鲁胺(Bicalutamide)。它们均为苯胺的衍生物,通过与内源性类固醇竞争并抑制AR,由于它们对于孕激素和糖皮质激素受体的亲和力有限,被认为是一种“纯”AR受体抑制剂[8]。
2.1.1 氟他胺(Flutamide)
氟他胺(图1A)是FDA于1989年10月批准的第一个非甾体AR抑制剂,它是第一代NSARI的基础。血浆半衰期约为6-8h。人血液中,2-羟基氟他胺是氟他胺的主要代谢产物,也是其主要活性形式,其血药浓度远高于氟他胺原药[8]。氟他胺单药治疗通常耐受性良好,并且在至少80%的患者中具有保持性欲和性潜能的作用(与醋酸环丙孕酮相比);氟他胺与LHRH激动剂联合用药可用于转移性PCa的初始治疗。氟他胺的主要不良反应是腹泻,肝毒性是限制其长期临床使用的主要原因[11-12]。
2.1.2 尼鲁他胺(Nilutamide)
尼鲁他胺(图1B) 是FDA批准的第二个非甾体AR抑制剂,其结构与氟他胺类似,但是半衰期较长(2d)。尼鲁他胺在体外与AR结合较弱,但在体内却能够产生持续的受体抑制作用。研究表明,尼鲁他胺除了抑制AR受体之外,还抑制雄激素的合成,特别是肾上腺来源的雄激素。重要的是,它不会损害糖皮质激素的合成。尼鲁他胺不可单独用药,必须与ADT相结合,其潜在的不良反应包括视觉障碍、酒精不耐受、性功能障碍和间质性肺炎[12]。
2.1.3 比卡鲁胺(Bicalutamide)
比卡鲁胺(图1C)是FDA批准的最广泛使用和研究的第一代NSARI,其结构与氟他胺类似,但是半衰期较长(7d)[8]。比卡鲁胺与大鼠AR的亲和力是氟他胺的4倍,在犬体内的ED50是氟他胺的50倍[13]。比卡鲁胺通过刺激DNA上转录不活跃受体的组装而发挥抑制AR的作用。比卡鲁胺可单一用药,与氟他胺相比,腹泻的发生率更低[14]。

图1 第一代NSARI结构式A:Flutamide;B:Nilutamide;C:Bicalutamide

图2 第二代NSARI结构A:Enzalutamide;B:Apalutamide;C:Darolutamide
2.2 第二代NSARI
第一代NSARI与AR-LBD亲和力较低。在CRPC患者中,长期服用时易出现抑制剂转化为激动剂的现象,出现“抗雄激素戒断综合征”。因此,第一代NSARI对去势抵抗性前列腺癌(CPRC)无效[15]。第二代NSARI直接靶向ARLBD并抑制受体复合物的核移位。目前已上市的药物包括恩扎鲁胺(Enzalutamide)、阿帕鲁胺(Apalutamide)和Nubeqa(Darolutamide)。
2.2.1 恩扎鲁胺(Enzalutamide)
恩扎鲁胺(图2A)是FDA于2012年8月批准的首个第二代NSARI,也是首个获FDA批准用于治疗非转移性去势抵抗性前列腺癌(nmCRPC)、转移性去势抵抗性前列腺癌转移性(mCRPC)、转移性去势敏感性前列腺癌(mHSPC)三种独特类型晚期PCa的药品。恩扎鲁胺对AR的亲和力明显高于第一代NSARI,AR的亲和力是比卡鲁胺的8倍[6]。尽管恩扎鲁胺与比卡鲁胺具有相同的药效基团,但它对AR靶基因无激动活性,能够抑制AR过表达的VCaP细胞(LNCaP/AR)生长并诱导其凋亡[16]。同时,恩扎鲁胺对突变的AR(包括比卡鲁胺耐药的AR-W741C突变)显示出抑制作用[17]。在LNCaP/AR CRPC小鼠异种移植模型中,与比卡鲁胺相比,恩扎鲁胺诱导肿瘤体积剂量依赖性的减小,该结果可以解释III期临床试验中,恩扎鲁胺治疗组患者总生存率增加28.9%(NCT00974311)的结果[18]。2019年,FDA批准了恩杂鲁胺的新适应症用于mHSPC。该批准是基于一项ARCHES III期临床试验(NCT00309985)。与安慰剂组相比,恩杂鲁胺组显著改善了影像学无进展生存期(rPFS),影像学进展或死亡风险显著降低61%[19]。值得注意的是,恩杂鲁胺潜在的不良反应是癫痫发作,其发生机制可能与恩扎鲁胺抑制脑内γ-氨基丁酸(GABA)受体相关[20]。因此,恩杂鲁胺禁用于有癫痫病史或有癫痫发作风险的患者[21]。
2.2.2 阿帕鲁胺(Apalutamide)
阿帕鲁胺(图2B)是FDA首个依据无转移生存期(MFS)的临床终点批准上市的NSARI,用于nmCRPC。阿帕鲁胺的结构和机制与恩杂鲁胺相似,在AR表达增加的情况下能够保持完全拮抗活性[22]。在体外,阿帕鲁胺与恩扎鲁胺具有相似的活性,但在CRPC的小鼠异种移植模型中,阿帕鲁胺在单位剂量下能够维持较高的稳态血浆浓度,显示出比恩杂鲁胺更强的肿瘤抑制作用[23]。阿帕鲁胺对GABA的亲和力较低,脑内含量比恩杂鲁胺低4倍。因此,阿帕鲁胺的中枢神经系统毒性和致癫痫潜能较低[24-25]。2019年,FDA批准了阿帕鲁胺的新适应症用于mHSPC。该批准是基于一项TITAN III期临床试验(NCT02489318)。与安慰剂组相比,阿帕鲁胺组显著改善患者24个月的影像学无进展生存期(68.2% vs 47.5%),影像学进展或死亡风险显著降低52%。阿帕鲁胺组24个月的总生存率也高于安慰剂组(82.4% vs 73.5%)。阿帕鲁胺组不良反应事件发生率略高于安慰剂组(42.2% vs 40.8%),其中皮疹更为常见[26]。
2.2.3 Darolutamide
Darolutamide(图2C)是FDA于2019年7月批准的用于治疗nmCRPC的NSARI。Darolutamide主要是通过阻断AR核转运从而抑制AR功能,对于AR的抑制作用明显强于恩杂鲁胺和阿帕鲁胺[27]。Darolutamide对耐AR受体突变的前列腺癌具有拮抗作用,如AR-F876L突变(恩扎鲁胺和阿帕鲁胺的耐药)、AR-W741L突变和AR-T877A突变(比卡鲁胺和氟他胺的耐药),但在AR-T878A突变时,其拮抗活性降低[28]。在细胞实验中,darolutamide抑制VCaP和LAPC-4细胞增殖,但对AR阴性细胞系、DU-145前列腺癌细胞和H1581肺癌细胞的增殖基本没有影响,证实darolutamide的抗增殖特性是AR依赖的前列腺癌细胞特有的[29]。在CRPC小鼠异种移植瘤模型中,darolutamide抑制VCaP/AR异种移植瘤生长,作用强于其他第二代AR抑制剂,而且不易通过血脑屏障,不会增加小鼠的血清睾酮水平[30]。临床研究显示,darolutamide能够抑制CRPC患者血浆中AR突变体的转录活性,特别是显著抑制了将恩杂鲁胺转化为部分激动剂的F877L、H875Y/T878A、F877L/T878A和先前未报道的T878G的转录活性[30]。在最新的ARADES III期临床试验(NCT02200614)中,与安慰剂相比,darolutamide显著延长患者MFS (40.4 vs 18.4个月,中位MFS总体改善为22个月)。患者总生存率增加,转移或死亡风险降低了59%。darolutamide与安慰剂组不良反应发生率相似,与安慰剂组相比,darolutamide与癫痫,骨折,认知障碍或高血压的高发生率无关[31]。目前,darolutamide正在进行mHSPC的III期临床研。
2.3 当前临床研究的NSARI
随着AR 抑制剂设计与研发的深入,特异、高效的NSARI显现出重要的临床意义。目前已有多个AR 抑制剂进入临床开发阶段。
2.3.1 GT-0918
GT-0918(图3A)是江苏开拓药业股份有限公司自主研发的AR受体抑制剂,处于临床III期试验阶段。GT-0918对AR具有高亲和力与高选择性,与恩扎鲁胺和阿帕鲁胺相比,对AR的亲和力增强[17]。此外GT-0918不仅作为AR的拮抗剂,而且可以下调AR蛋白的表达。因此,GT-0918抗CRPC的作用普遍强于现有上市的NSARI。GT-0918在动物的中枢神经系统分布较少,诱发癫痫的风险更低[12]。目前,GT-0918的2项临床试验已在clinical trials.gov上注册,旨在评估评估GT-0918对于mCRPC患者的安全性和有效性(NCT02826772,NCT03899467)。
2.3.2 HC-1119
HC-1119是成都海创药业有限公司自主研发的第二代NSARI,属于恩杂鲁胺的氘化类似物,处于临床III期试验阶段。HC-1119改变了恩杂鲁胺的PK性质,提高了血浆药物暴露量(抗肿瘤活性是恩扎鲁胺的2倍),降低了脑内暴露量。因此,HC-1119的诱发癫痫的可能性较低[32]。目前,HC-1119的5项临床试验已在clinical trials.gov上注册,其中2项研究是III期临床试验,旨在评估口服HC-1119与安慰剂(NCT03851640)或口服恩杂鲁胺(NCT03850795)相比对mCRPC患者的疗效和安全性。值得注意的是,使用恩杂鲁胺作为对照药物能够更加直观的比较氘代药物的药效与安全性。据报道,预计2021年完成III期临床研究[33]。
2.3.3 SHR-3680
SHR-3680是恒瑞医药自主研发的AR受体抑制剂,处于临床III期试验阶段。与第一代AR抑制剂(如比卡鲁胺)不同,SHR3680是AR的完全拮抗剂,通过与AR竞争性结合,抑制AR核转移,抑制AR与DNA结合,最终抑制AR靶基因的转录。目前,SHR-3680的4项关于前列腺癌的临床试验已在clinical trials.gov上注册,其中1项为III期临床试验,旨在比较SHR-3680与比卡鲁胺治疗激素敏感型前列腺癌的安全性和有效性( NCT03520478)。
2.3.4 ODM-204
ODM-204(图3B)是 由Orion Corporation研 发 的 对AR转录和CYP17A1具有双重抑制活性的化合物,处于临床II期试验阶段。ODM-204对于CYP17A1的抑制活性类似于Galeterone,AR抑制作用类似于恩杂鲁胺[17]。目前,ODM-204的1项临床试验已在clinical trials.gov上注册,旨在评估评估ODM-204在mCPRC患者中的安全性、耐受性和药代动力学特性(NCT02344017)。
2.3.5 EPI-001
EPI-001(图3C)是由Essa Pharma研发的一种独特的AR抑制剂,处于临床II期试验阶段。EPI-001能够共价结合和抑制AR-NTD,靶向AR蛋白的N-末端克服目前AR靶向制剂的耐药性。EPI-001抑制AR依赖性LNCaP细胞的增殖,但不抑制AR 非依赖性PC3或DU145细胞的增殖。在去势的LNCaP CRPC小鼠异种移植瘤模型中,EPI-001给药组显著降低瘤体积[34]。
2.3.6 TRC-253
TRC-253(图3D)是 由Janssen Research & Development LLC研发的恩杂鲁胺类似物,处于临床II期试验阶段。TRC-253是一种高效、高亲和力的竞争性AR抑制剂,也是多种AR突变体的抑制剂,包括AR-F876L。TRC-253旨在通过特异性靶向AR-LBD中的突变体来解决当前AR抑制剂的耐药性,同时它还能够抑制野生型AR信号传导[17]。目前,TRC-253的1项临床试验已在clinical trials.gov上注册,用于mCPRC患者中剂量递增研究(NCT02987829)。
2.3.7 AZD-3514
AZD-3514(图3E) 是由AstraZeneca plc研发的AR抑制剂。AZD-3514不仅抑制AR的核移位,而且与GT-0918一样能够诱导AR表达下调。目前,AZD-3514已经完成了2项I期临床研究(NCT01351688,NCT01162395),它在临床研究中显示出良好的肿瘤抑制作用,但在转移性CRPC患者中仅报道了中度抗肿瘤活性[11]。
2.3.8 BMS-641988
BMS-641988(图3F)是 由Bristol-Myers Squibb Co公司研发的AR有效竞争性抑制剂。BMS-641988对AR的亲和力比比卡鲁胺高20倍,体外抗雄激素活性是比卡鲁胺的3-7倍,但是对于AR可能存在较弱的激动活性[12]。目前,BMS-641988已经完成2项I期临床研究(NCT00644488,NCT00326586),结果显示BMS-641988部分激动AR受体结果与临床前研究一致,在治疗剂量下抗肿瘤作用较弱,且诱导癫痫活性较强,导致其研究终止[35]。
2.3.9 ONC1-0013B
ONC1-0013B(图3G)是由Avionco LLC公司研发的恩杂鲁胺类似物。ONC1-0013B能够有效抑制DHT刺激的前列腺癌细胞PSA的表达和增殖,防止雄激素与AR配体结合区结合,抑制AR核移位和抑制辅活化子复合物的形成。在前列腺癌LNCaP-Z2异种移植模型中,ONC1-0013B抑制肿瘤生长并抑制PSA表达。在相同剂量下,ONC1-0013B的体内活性与恩杂鲁胺相当。ONC1-0013B在脑内的分布低于阿帕鲁胺,GABA相关癫痫发作的风险较低[36]。目前,ONC1-0013B已经完成1项I期临床研究(NCT03074032),结果未见报道。
2.4 新发现的NSARI
IMTPPE与JJ-450
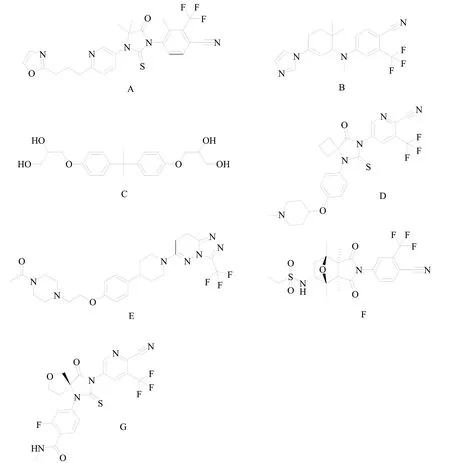
图3 当前临床研究的NSARI结构式A:GT-0918;B:ODM-204;C:EPI-001;D:TRC-253;E:AZD-3514;F:BMS-641988;G:ONC1-0013B;

图4 新发现的NSARI结构式A:IMTPPE;B:(-)-JJ-450;C:(+)-JJ-450
Yang等[37]通过高通量筛选得到了一种新的AR拮抗剂IMTPPE(图4A),能够抑制CRPC细胞(包括对恩杂鲁胺耐药的细胞)中的核AR水平和活性。同时发现,IMTPPE的类似物JJ-450(图4B-C)对AR转录活性的直接和特异性抑制作用。JJ-450抑制AR募集,抑制AR靶基因的表达。重要的是,JJ-450抑制AR-V7转录活性及其靶基因表达,包括表达AR-V7的22Rv1细胞。这些发现表明JJ-450是一类新的AR拮抗剂,对CRPC具有治疗潜力,包括对恩杂鲁胺耐药癌细胞。
3 总结与展望
前列腺癌是男性患者患病风险最高的癌症之一,几十年来一直有靶向治疗选择,主要目的是抑制AR。在AR抑制剂中NSARI表现出更好的临床适用性,近30年来世界范围内已批准多个NSARI用于前列癌治疗。经过多年的临床应用,尽管这类药物大都已被证实具有优异的临床治疗效果,但随着临床研究的深入多数药物均表现出不同程度的耐药性。这可以从讨论AR、耐药性、抗雄激素和激动剂效应的文章数量的急剧增加中看出。因此,基于对现有NSARI耐药机制的研究或结构优化的基础上,一些新颖结构的NSARI被设计出来,这些化合物的临床前或者临床研究结果表明他们可以一定程度克服现有NSARI的缺陷。新一代NSARI有望改善前列腺癌标准疗法,减少副作用。因此,使用AR抑制剂作为一种治疗策略,通过增加治疗作用来克服对于初级治疗的抵抗,变得越来越有吸引力。此外,最新的研究表明,新的AR抑制剂药物并没有完全取代一些旧的标准。目前,一些关于优化利用现有雄激素靶向药物治疗顺序与组合的试验正在开展,激素和化疗药物进行比较和/或联合使用至关重要,相关研究的结果值得肯定,并有望帮助指导未来的个性化治疗。随着NSARI治疗前列癌研究的深入,通过新型NSARI的设计、对已上市NSARI新的临床适应症的发掘和精确定位结合NSARI联合应用,对于解决当前上市NSARI在临床应用过程中所发现的和不良反应具有重要意义。
