PO分子在外电场下的特性研究
2020-12-05梁冬梅孙光宇
梁冬梅, 荆 涛, 孙光宇
(1.凯里学院理学院,贵州凯里 556011;2.贵州师范学院物理与电子工程学院,贵州贵阳 550018)
磷或含磷的化合物与氧反应,可以生成各种磷氧(PxOy)化合物,比如,P2O5,PO3,P2O4,P2O,PO2等,PO分子是其中一种磷和氧反应的生成物.一直以来,PO分子的研究受到人们的广泛关注[1-6].早在1984年,Lawrence[7]从理论上研究了气体氧化物PO和PO2.自此以后,许多技术和方法被应用到磷化合物的研究中[8-10].2003年,Yahia[11]等从实验上研究了PO分子电子基态的阳离子和阴离子的特性,并与理论数据进行对比.2019年,韩晓琴[12]等采用从头算方法研究了PO分子的光谱常数和势能曲线.从近年来的文献看,对外电场下PO分子的性质还没被研究过.而在外场作用下分子会出现新变化和新现象[13],这可为进一步研究此领域提供参考.
图1 PO的分子结构
在没有外电场作用下,图1给出了PO分子的结构.为了使所得结果更加可靠和更有说服力,在文献[12]计算的基础上,本文使用与其相同的基组进行下面的计算,即:对PO分子在Z轴方向上依次加上-0.04~0.04a.u.的外电场,采用B3LYP方法、以6-311G(3df)为基组进行结构优化.优化后使用相同的基组,在不同强度和方向的外电场下采用杂化CIS方法研究PO分子的激发能、激发波长和振子强度.这里,1a.u.=5.14225×1011V/m.
1 理论和计算方法
考虑微扰的作用,含时薛定谔方程可写为[14],

其中,H0是无外场作用时的哈密顿量,H′(t)是外场与体系作用的哈密顿量.
偶极近似下分子与外场的作用能可写为,

式中μ为分子的电偶极矩.
根据Grozema[15,16]等所提出的模型,沿Z轴方向对PO分子结构在-0.04~0.04 a.u.的外电场下进行优化,并进一步采用CIS-B3LYP/6-311G(3df)方法对PO分子的激发态物理量,包括激发能、激发波长和振子强度等进行了计算.
2 结果分析
2.1 外电场下PO分子的结构和电荷分布
使用密度泛函理论(Density Functional Theory,DFT)中的B3LYP/6-311G(3df)方法,沿Z轴方向优化了不同外电场(包括 -0.04、-0.03、-0.02、-0.01、0.01、0.02、0.03、0.04 a.u.)下PO的分子结构,表1列出了优化后分子的基态结构参数.
从表1可以看出,PO分子的键长随外电场的增大发生了明显的变化.在没有外电场存在时,键长R=0.14759 nm,这与文献[12]和实验[17]的结果是一致的.随着正向电场的不断增大,键长Re不断减少,在F=0.04a.u.时,键长R的数值达到最小,为0.14629 nm,如图2所示.从图3可以看出,PO分子体系的总能量随着电场从-0.04a.u.变化到0而不断增大,当F=0时,总能量达到最大值-416.59937 a.u.,随后,总能量随着电场从0增大到0.04 a.u.而不断减少.如图4所示,分子偶极矩μ随电场从-0.04a.u.变化到0.04 a.u.而不断减小,从1.9906减小到1.8348 Debye,减小了0.1558 Debye,这说明外电场对PO分子的极性影响不大.

表1 不同外电场下优化的PO分子的键长、总能量和偶极矩

图2 键长R随电场的变化
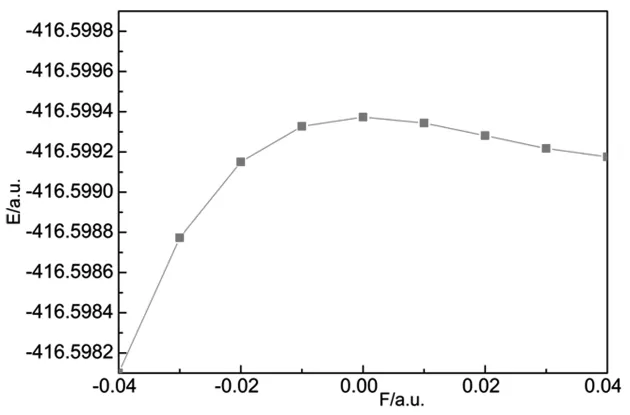
图3 分子总能量E随电场的变化
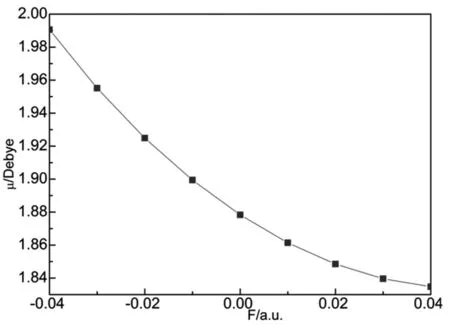
图4 偶极矩μ随电场的变化

图5 HOMO、LUMO随电场的变化

表2 不同电场下PO分子的电荷分布
从表2可以看出,PO分子的电荷分布依赖于外电场的分布.磷原子(P)带正电荷,呈电正性,氧原子(O)带负电荷,呈电负性.由于分子本身不显电性,所以P原子所带正电荷数和O原子所带负电荷数的总和为零.当电场从-0.04变化到0.04 a.u.时,P原子周围的正电荷密度从0.461471减小到0.450393,O原子周围的负电荷密度由开始的-0.461471变为最后的-0.450393.分子的稳定构型在外电场作用下是由分子所受的外电场力和分子的内应力的合力所决定,电子带负电,在电场的作用下会逆着电场方向进行移动.随着外电场的增大,离子间库仑力变小,使得P-O之间的电场减弱,内应力大于外电场力,分子键长变小,总能量增大,偶极矩减少.
2.2 外电场下PO分子的能级变化
使用B3LYP/6-311G(3df)方法,沿图1所示的Z轴方向优化了不同外电场下PO的分子基态构型,得到如表3和表4所示PO分子α电子和β电子的最高占据轨道HOMO、最低空轨道LUMO和能隙Eg,如图5所示.最高占据轨道反映了分子丢失电子的能力,最低空轨道反映了电子亲和力的大小.从图5可以看出,随着正向外电场强度的不断增加,α电子的HOMO轨道和LUMO轨道的能量均不断增大,由能隙Eg=LUMO-HOMO可以得出,能隙在外电场作用下不断减小,但减小的幅度并不大.而β电子的最高占据轨道HOMO不断减小,LUMO轨道的能量不断增大,能隙Eg不断增大,虽然增大的幅度并不大,但整体上高于α电子的能隙值.这表明α电子比β电子更容易受外电场的影响,从HOMO轨道跃迁到LUMO轨道,形成空穴.
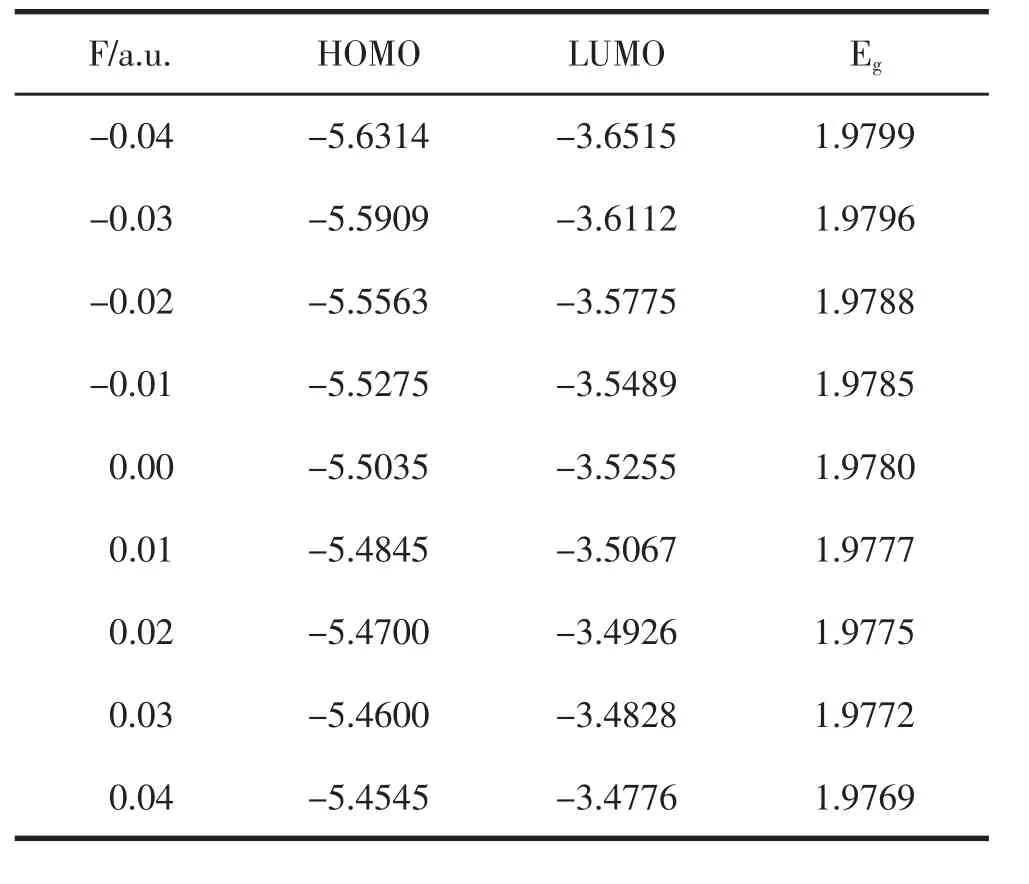
表3 外场下HOMO、LUMO和能隙Eg变化(α电子)(单位:eV)
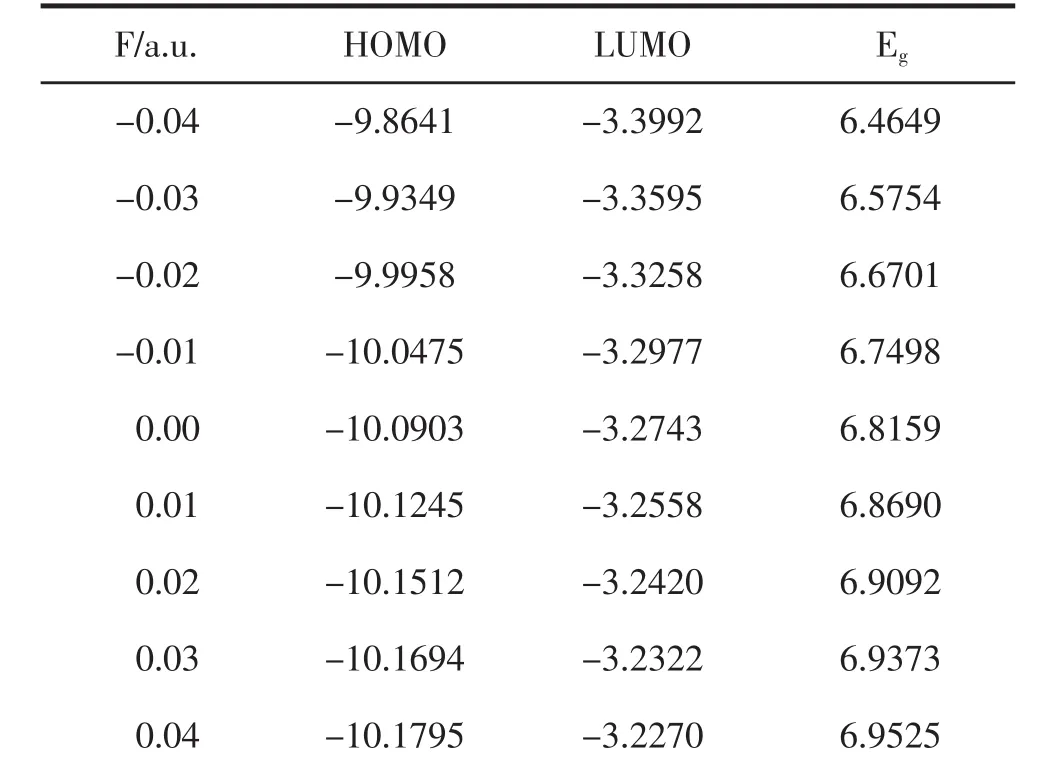
表4 外电场下HOMO、LUMO和能隙Eg变化(β电子)(单位:eV)
2.3 外电场下PO分子的激发态
在优化后得到分子稳定构型的基础上,使用CIS-B3LYP/6-311G(3df)方法,在不同强度外电场下研究了PO分子的激发态能量E、波长λ和振子强度f,结果如表5至表7所示.
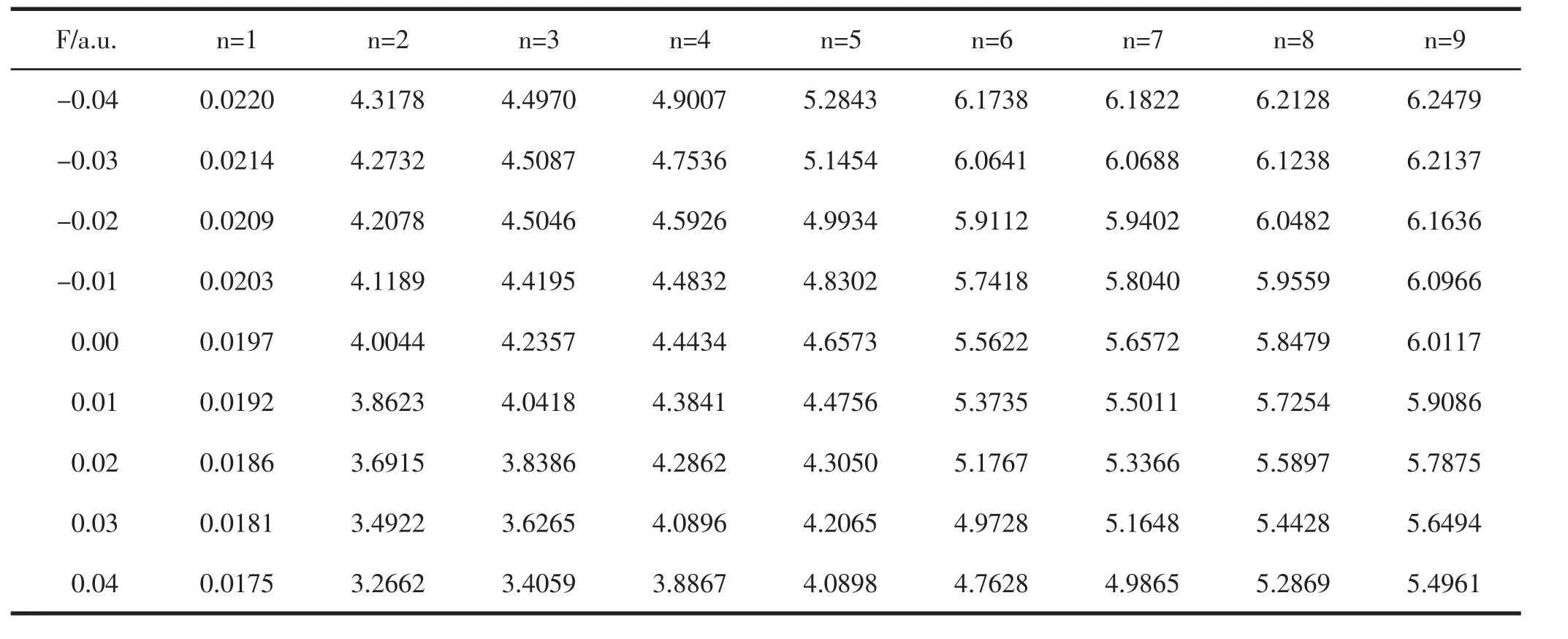
表5 不同外电场下PO分子激发能E的变(单位:eV)
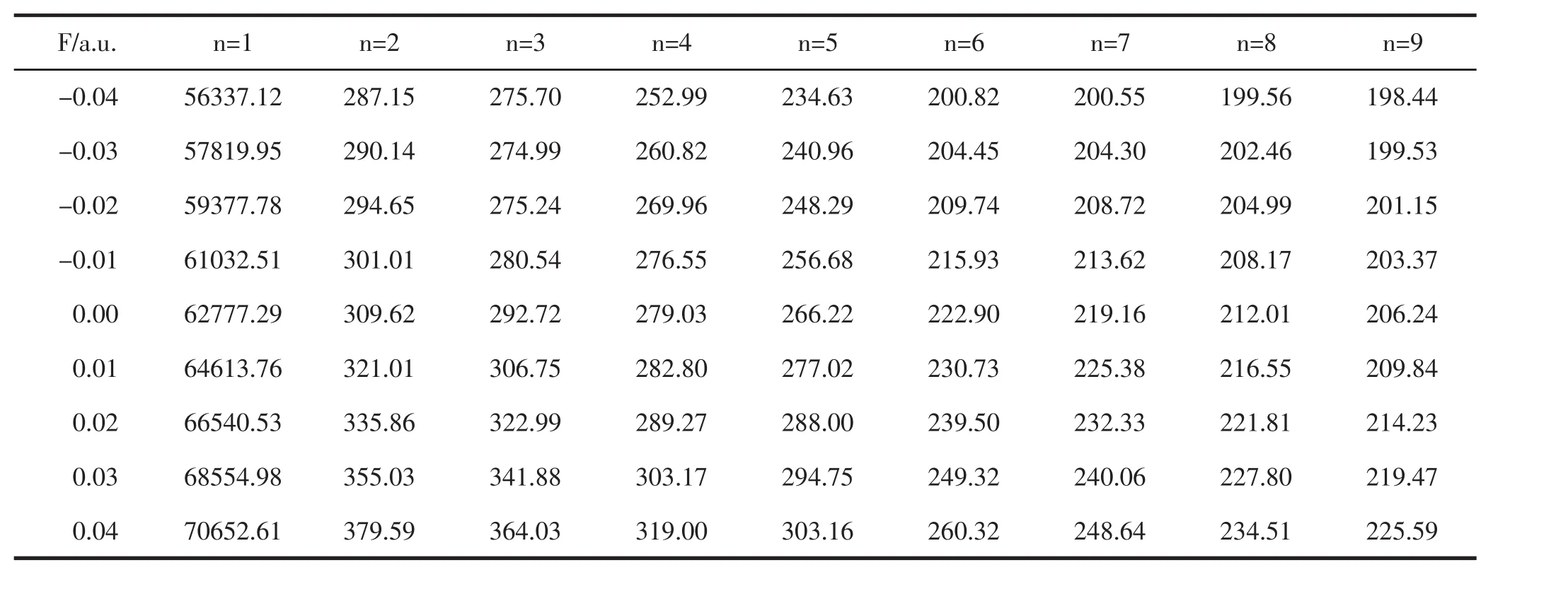
表6 不同外电场下PO分子激发波长λ的变化(单位:nm)
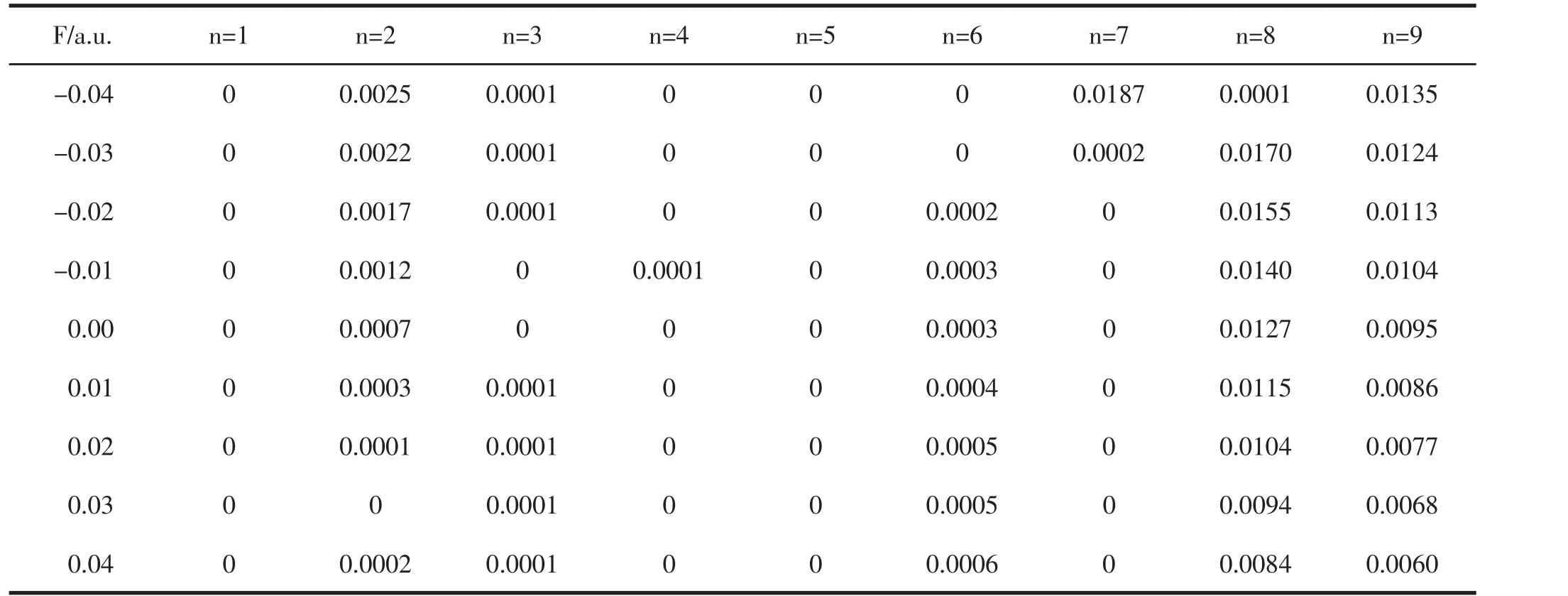
表7 不同外电场下PO分子振子强度f的变化
从表5和表6可以看出,随着外电场从-0.04变化到0.04 a.u.,各激发态的能量E不断减小,这表明分子激发变得越来越容易,而各个激发态的激发波长随正向电场的增加不断增大,但并没有在可见光区出现.振子强度为零,则该跃迁属于禁阻跃迁.从表7可看出,外电场的引入对原来电子的跃迁几乎没有影响.原来的禁阻跃迁在加入外电场后,其振子强度仍为零,并没成为可允许的跃迁.有的激发态的振子强度虽不为零,但强度很小,在实验上观察不到,如第八激发态和第九激发态.总之,外电场虽然对该分子的激发能和激发波长产生了显著影响,但并没有改变分子的跃迁特性.
3 结论
本文对PO分子不同外电场下的构型采用B3LYP/6-311G(3df)方法进行了优化,并对该分子的基态结构参数,包括总能量、偶极矩和电荷分布等随外电场的变化进行了讨论.在此基础上,进一步采用CIS-DFT方法研究了不同外电场下PO分子的激发态变化.随着电场从-0.04变化到0.04 a.u.,PO分子的键长不断减小,总能量呈先增加后减少的变化,偶极矩在电场作用下逐渐减小.激发能、激发波长随外电场的增加变化明显,而振子强度几乎没有随外电场的改变而改变.
