活性天然产物蛋白靶点的快速筛选策略
2020-12-04陈鹏
陈鹏
(中国中医科学院医学实验中心,北京 100700)
活性天然产物是来源于自然界中具有生物活性和药用潜力的化学物质[1-2]。从意外发现青霉素到从柳树皮中提取的阿司匹林[3-4],历史上天然产物及其衍生物占创新药物总数的一半以上[5]。中药是丰富的天然产物资源库,从青蒿中提取的青蒿素直到今天仍是全球抗击疟疾的重要武器[6],从砒霜得到的砷制剂成为治疗白血病的良药[7]。
现代药物的研发通常从靶点出发,经过计算机设计,合成,筛选,药效验证,具有较明确的药理毒理说明[8],而活性天然产物的筛选和使用往往基于实际药效筛选,在成药过程中需要对其药理和毒理进行补充性研究[9]。蛋白质是细胞功能的主要行使者,细胞依托于蛋白质的功能完成生命周期中的各种生命活动。药物的重要作用途径是与蛋白质等大分子相互作用对细胞生命活动完成调控[10-11]。筛选和揭示天然药物的靶点,是天然产物成药过程中药理和毒理解释不可或缺的环节[12-14]。药用植物资源的提取利用是提高药用植物资源附加值的重要方式,中药的科学内涵需要进行深入研究和阐释,寻找和鉴定中药的药物靶点特别是复方制剂,是理解其药理和有效物质基础的关键步骤[15-17]。在对抗新型冠状病毒引起的疫情(Coronavirus Disease 2019,COVID-19)中,一些中药制剂展现了良好的疗效[18],明确其药物靶点能够为其在国际推广提供科学依据。
传统的天然产物的蛋白靶点发现是利用探针标记的方法对天然产物进行示踪[19],对靶点蛋白进行亲和纯化后进行鉴定,不论是早期简单的标记生物素、微球还是后来发展的柔性探针,活性探针,光催化探针等都无法避免固有的缺陷,首先探针合成依赖于专业的化学平台,合成和表征的时间长,延长了药物研发周期,其次天然产物的标记大多会改变其原有的结构,影响其活性[20-22]。针对以上问题,近年来发展了各种基于非标记的快速天然产物靶点筛选技术。这些技术整体上可以归纳为两大类,一类是利用天然产物自身以及与靶点互作的理化性质进行直接筛选;另一类是利用基因组文库或者生命体宏观表型变化对靶点进行间接筛选。本文从以上角度综合靶点验证的技术对目前活性天然药物的快速筛选方法进行综述,以期给读者带来有益参考。
1 活性天然产物蛋白靶点的直接筛选策略
1.1 基于天然产物自身理化性质的筛选策略
天然产物结构多样,含有共轭结构体系的分子往往具有紫外吸收,测定紫外吸收强度是这类物质检测的常用技术手段[23-24],同时大共轭体系、分子环化以及苯环等官能团的存在使得一部分天然产物具有荧光属性,在一定波长的激发光下可以自发荧光,如白藜芦醇等物质具有一定的自发荧光[25],这些特殊理化性质的存在使得在筛选此类药物的靶点时带来便利。利用药物的自发荧光可以研究细胞对药物的利用吸收[26],药物的细胞定位,结合凝胶电泳和质谱技术可以方便快速的找到药物结合的蛋白靶点,进而进行分子机制的研究。此类方法只局限于部分天然药物,很多具有重要价值的药物没有明显的紫外吸收和荧光光谱,比如青蒿素[27]。
1.2 基于天然产物靶点蛋白质稳定性的直接筛选
通常小分子药物与靶蛋白的结合会增加蛋白的稳定性,研究人员利用药物小分子对蛋白质氧化、酶解、加热过程中稳定性的影响,结合蛋白质组学技术开发了一系列药物靶点的非标记快速筛选方法[28]。
1.2.1 基于靶点氧化稳定性开发的SPROX技术基于氧化稳定性结合定量质谱蛋白质组学,研究人员构建了SPROX(Stability of Proteins from Rates of Oxidation)技术,这种技术以不同浓度盐酸胍处理混合蛋白(细胞组织裂解液),然后用同样条件的H2O2处理,用蛋白质组学定量氧化的甲硫氨酸的氧化比例,药物的加入会使得靶点蛋白质结构稳定性增加,进而减少甲硫氨酸暴露和氧化。作者以免疫抑制剂环孢素A(Cyclosporin A,CsA)和酵母蛋白为例进行了研究,发现CsA能显著降低10种蛋白质中甲硫氨酸的氧化比例,其中cyclophilin A和UDPglucose-4-epimerase是已经发现的两种效应靶点,另外8个靶点同时为研究该药的脱靶效应提供了思路,同时利用该方法还能够对天然产物与靶点蛋白的结合亲和力进行测定[29]。
Geer Wallace等[30]利用该技术结合标记定量蛋白质组学研究了天然产物manassantin A潜在作用靶点,该技术也可以研究生命过程中蛋白质稳定性的差异,蛋白质结构稳定性是近年抗衰老领域的研究热点,Roberts等[31]利用该技术研究了衰老模型小鼠与青年小鼠脑中蛋白质稳定性的差异,为寻找抗衰老的蛋白质治疗靶点提供了示范。
1.2.2 基于靶点酶解稳定性开发的DARTS技术 DARTS(Drug affinity responsive target stability),是基于药物亲和反应提高靶点蛋白抗蛋白酶水解的特性开发的新技术。Lomenick等[32]首先利用小分子雷帕霉素、FK506 抑制剂和其重组表达的靶蛋白FKBP12进行了原理验证,实验结果表明加入与该蛋白结合的小分子可显著提高其抗蛋白酶水解的特性,且与小分子浓度呈依赖性,同时mTOR激酶与其抑制剂E4的相互作用也验证了上述原理。该团队运用该技术进行实践,成功的筛选出了白藜芦醇调控真核转录的靶点蛋白EIF4A,Lomenick认为该方法不仅仅体现在对非标记小分子产物靶点筛选得适用,而且也可以在生物分子相互作用筛选之间广泛适用[32-34],近年来该方法在天然产物应用领域应用比较广泛[35]。该技术也具有一定的缺陷,酶解法对低丰度的蛋白损失严重,裂解液使用具有限制,获得的靶点倾向于高丰度的蛋白。小分子本身对酶活的影响以及复杂的蛋白间相互作用也可能带来结果的偏差,同时该方法也受凝胶、液相分离和质谱仪器的检测限的影响[20]。
1.2.3 基于靶点热稳定性开发的CETSA/TS-FITGE/TPP/ITSP技术 基于靶点热稳定性开发的靶点筛选技术是近几年研究的热点,实验证实大部分情况下小分子和其靶点作用能够增加靶点的稳定性,同样靶点蛋白质随温度变化的变性曲线会因为与配体小分子的结合而变化,靶点蛋白的降解温度和变化趋势可以作为区分靶点蛋白和其他蛋白的一个参数,进而完成靶点蛋白的筛选工作[36-38]。相比于氧化稳定性和酶解稳定性,靶点蛋白热稳定性无需化学试剂的添加,而是直接采用加热超速离心,结合分离定量等技术测定对照组和药物干预组可溶性蛋白的差异[39]。该原理基于不同的蛋白质定量差异分析方法,形成了不同的细分技术种类。
研究人员将热稳定性与凝胶电泳和免疫印迹结合对药物特异蛋白靶点结合进行了分析,建立为细胞热稳定性迁移试验CETSA(Cellular Thermal Shift Assay)技术,在温度和加热时间保持恒定的情况下,可获得与药物浓度依赖性的可溶性靶点蛋白特异性图谱,这种方法依赖于特异性的抗体,因此通量较低[40]。另一组研究人员将荧光定量二维凝胶电泳蛋白质组技术与热稳定性原理结合起来,绘制药物干预下蛋白的热稳定性迁移曲线,开发出基于凝胶的蛋白质组学筛选药物靶点的新方法TS-FITGE(In-gel fluorescence difference caused by thermal stability shift),成功找到了大麦芽碱(Hordenine)新的药物靶点并进行了体外验证[41]。但无论是普通的还是半定量的荧光染色差异凝胶二维电泳,其分辨率都有限。
研究人员将热稳定性与定量质谱蛋白质组学技术联合运用在建立了TPP(Thermal proteome profiling)方法,在这项工作中细胞裂解液全蛋白在与广谱激酶抑制剂staurosporine反应,然后在不同温度下加热,使用基于质谱的工作流程从每个样品中提取、鉴定和定量可溶性蛋白质。利用这种方法作者分析了激酶抑制剂staurosporine对其50多个靶点的影响,鉴定出血红素生物合成酶铁螯合酶作为该激酶抑制剂的新靶点,并认为它的抑制作用导致了使用 vemurafenib和alectinib观察到的光毒性,同时还观察到该药物治疗下游通路蛋白的热迁移[42]。利用该技术还发现methotrexate、(S)-crizotinib 和2'3'-cGAMP的细胞内作用靶点,其中发现2'3'-cGAMP与先天免疫信号传导的跨膜受体STRING蛋白相互作用[43]。
随后研究人员对该方法进行了更加详细的描述,并对裂解液,离心条件,蛋白质酶切条件,质谱数据分析流程等进行了系统的优化[44-45]。基于靶点热稳定性的技术也不可避免的存在一些缺陷,比如低丰度蛋白易降解难以检测,产生的大量数据难以分析,药物对热稳定性的影响具有不确定性以及间接作用的影响带来假阳性。Ball等[46]提出TPP技术温度设置点过多,操作繁琐成本高,他们开发了基于单一温度的热稳定性靶点筛选方法iTSA(Isothermal shift assay),该技术操作简单,理想的情况下效率更高。
1.2.4 聚合物捕获法 特异性聚合物捕获法UPT(Unique Polymer Technology)不同于基于靶点稳定性的方法,研究人员合成了一些特异的高分子聚合物材料,这些材料能够非共价结合小分子药物,进而捕获与小分子结合的蛋白靶点。研究人员将双吲哚甲酰胺III(GSK3β的抑制剂)固定在聚合物表面,用免疫印迹分析方法证明了GSK3β的特异性捕获[47]。UPT的方法显然相对于以上基于稳定性的方法操作更加简单[48],该方法的局限性是不仅仅把与小分子相互作用的靶蛋白富集出来,而且还会富集与靶蛋白相互作用的其他蛋白质,假阳性率较高[48]。
2 天然产物靶点间接筛选方法
2.1 基因组文库筛选
基因组文库筛选是通过分子生物学方法得到基因敲除、表达量降低、过表达、突变的个体或者细胞,并将含有以上不同基因的模式生物个体或者细胞集合成文库,进行高通量药物靶点筛选的方法。利用基因文库对研究对象-天然产物进行药效学评价,产生药性改变的对象可能就是敲除了天然药物对应的靶点,例如在敲除掉特定基因的细胞如果产生了对药物的耐药性,那么该基因编码的蛋白就有可能是药物的靶点。基因文库构建的方法主要有RNAi,同源重组,基因编辑等方法[49]。酵母是一种应用广泛具有价值的研究疾病和药物作用机制的模式生物,拥有遗传易感,稳定,培养简单的特点[50]。基于酵母建立的基因缺失、过表达的文库已经广泛应用于筛选药物的靶标[51]。最近,基于基因编辑技术CRISPR(Clustered regularly interspaced short palindromic repeats)-Cas9技术获取的真核细胞文库也已经构建和应用[52-53]。
2.2 其他间接筛选方法
除了通过天然产物小分子与靶点蛋白结合后理化性质的改变作为筛选方法,也可以间接的通过药物刺激后观察细胞生理、信号通路蛋白或者基因水平的变化,进而利用功能分析和生物信息学辅助判断可能的靶点蛋白。例如,活性产物小分子对血管生成产生影响、诱导自噬等现象[54-55],就可以推断分子可能的作用靶点范围,进而利用分子生物学方法逐一验证,针对虎杖苷诱导肿瘤细胞产生氧化应激的现象,结合蛋白质组学分析,发现虎杖苷是一种新的葡萄糖-6-磷酸脱氢酶的抑制剂[56]。雷公藤甲素表现出强烈转录抑制现象,根据这一现象研究人员借助中心法则原理,推导并验证出RNA聚合酶Ⅱ的亚基XPB是雷公藤甲素的靶点,解决了困扰科研人员多年的问题[57]。值得一提的是,无论是辅助组学技术进行候选靶点的筛选,还是计算预测可能的结合位点,生物信息学都深度参与了整个研究过程,随着人工智能的发展,这一趋势将更明显,甚至起到决定性作用[58-59]。
3 天然活性产物靶点确证方法
在筛选得到天然产物的候选靶点后,需要进一步的进行确证,直接的方法就是采用与筛选方法一致的技术,如可以通过靶点的纯蛋白与小分子进行结合前后的稳定性测试,如果小分子与蛋白质之间是共价结合,还可以采用质谱的方法测量结合分子后蛋白的分子量,从而判断结合的小分子数目,甚至解析结合的位点[60-62]。除了这些常规的方法,还可以通过测量小分子与蛋白结合前后的理化参数来做出判断,常用的方法有:等温滴定量热(Isothermal titration calorimetry,ITC)、表面等离子体共振(Surface plasmon resonance,SPR)、光谱法、原子力显微镜、核磁共振法等[63-64]。
4 展望
中药以及活性天然产物源自长期的实践,具有真实可靠的疗效,筛选挖掘其药物靶点可能发现新的治疗靶点,同时新的药物靶点的发现也为天然药物的结构改造,设计新的药物提供了思路。如图1所示,药物与靶点就像太极图的阴阳互补的两面,“药-靶互动”的思想将会促进新药的创新与改造。
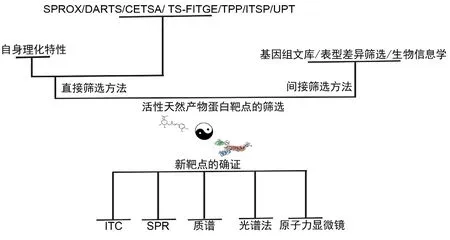
图1 活性天然产物蛋白靶点快速筛选与验证策略总结
目前针对复方中药复杂组分的药物靶点筛选,主要是利用生物信息学网络药理学进行的[65-67]。在COVID-2019疫情中也大量涌现了基于网络药理学的研究成果[68]。组分复杂的中药复合物是很难通过化学标记的方法进行分析,单个组分的研究也不可能解释中药复方多组分多靶点的优势,借助非标记化合物的蛋白质靶点新型筛选策略,从宏观角度研究中药复方的靶点群,可能会对中药的复方的分子机理解释、解决中药注射液不良反应等问题提供帮助。
中药的现代化需要科学的手段对中药的科学内涵进行阐释。寻找和鉴定中药的药物靶点,是理解中药药理和明确其有效物质基础的关键步骤。生物化学的发展给我们提供了有可能解决这些问题的技术手段。在COVID-2019新型冠状病毒引起的突发公共卫生事件中,一些老药、中药、天然产物显示出明显的疗效[69-70],快速的筛选这些药物的作用靶点,可以为这些药物的应用和推广提供科学依据和信服的实验药理证据。本文总结了活性天然产物效应靶点的快速筛选策略,探讨了各种技术的优缺点及前景,希望能够对相关研究提供新的思路。
