经典型21-羟化酶缺乏症患儿基因型预测临床分型的研究
2020-11-27兰天,姚辉
兰 天,姚 辉
0 引 言
先天性肾上腺皮质增生症是最常见的常染色体隐性遗传病之一,由CYP21A2基因突变所致的21-羟化酶缺乏症(21-hydroxylase deficiency,21-OHD)占先天性肾上腺皮质增生症的90%~95%。中国的发病率为1∶12 200~1∶16 466[1]。21-OHD临床分型为经典型和非经典型。经典型又可进一步分为单纯男性化型和失盐型。多项研究表明,CYP21A2基因型与21-OHD表型之间存在相关性[2-5],但不同种族间CYP21A2基因的突变频率差异较大[6-8]。随着21-OHD的新生儿筛查逐渐普及,近几年该病的诊治模式发生了变化,越来越多的患儿通过新生儿筛查被检出。患儿确诊时往往缺乏典型的临床表现而易误诊。新生儿筛查检出的21-OHD患者缺乏精确的临床分型,治疗时药物的种类和剂量也更难把握。本研究旨在探讨湖北地区经典型21-OHD患儿基因型与临床分型关系,通过基因型预测临床分型,为新生儿筛查阳性患儿的诊疗提供帮助。
1 资料与方法
1.1 研究对象回顾性分析2010年8月至2019年3月武汉儿童医院遗传代谢内分泌科64例确诊并入组的经典型21-OHD的患者临床资料。其中男38例,女26例,年龄7天~6.9岁(平均年龄1.4个月)。纳入标准:参考2016年《先天性肾上腺皮质增生症21-羟化酶缺陷诊治共识》[1];均为汉族,祖籍湖北。排除标准:由新生儿筛查阳性诊断的21-OHD,其他类型的先天性肾上腺皮质增生症,如3β-羟化酶缺乏症等。本研究获得武汉儿童医院伦理委员会的批准(批准号:武儿医2017020),患儿父母均签署了知情同意书。
1.2研究方法
1.2.1 资料收集记录经典型21-OHD患者的具体临床分型(失盐型、单纯男性化型)、基因检测结果,包括CYP21A2基因型、突变位点等。
1.2.2临床分型失盐型:出现恶心、呕吐、喂养困难、脱水、体重增长缓慢,伴有低血压、低钠高钾血症、17-羟孕酮水平升高、血浆肾素活性增加、皮质醇和醛固酮降低。单纯男性化型:女性外生殖器异常(阴蒂增大似阴茎、阴唇融合等)、男童性阴茎增大(但睾丸不增大)、阴毛早现、血清17-羟孕酮水平升高、身高增长速率过快、骨龄超前,早期身高高于同龄儿,随着骨骼的成熟最终导致身材矮小[1]。
1.2.3基因检测二代测序联合多重链接依赖探针扩增技术(multiple linked probe amplification,MLPA),外送金准基因检测公司。
1.2.4基因型分组根据21-羟化酶受损的严重程度,将突变从重到轻分为极重度突变组(n=15)、重度突变组(n=23)、中度突变组(n=23)、未知突变组(n=3)。极重度突变组包括Del、p.G110fs、Cluster6E (p.I236N、p.V238E、p.M240K)、p.Q319X、 p.R357W、p.484P的任意组合,可导致21-羟化酶活性完全丧失。重度突变组包括I2G的纯合突变、I2G与极重度突变组组成的复合杂合突变,会导致21-羟化酶活性小于正常的1%。中度突变组包括p.I173N的纯合突变、p.I173N与重度突变组、极重度突变组组成的复合杂合突变,21-羟化酶活性为正常酶活性的2%。未知突变组包含有罕见突变位点的纯合突变或复合杂合突变。由于未知突变组突变引起的21-羟化酶活性改变未知,因此不纳入基因型预测临床表型研究范围内。
1.3统计学分析采用SPSS 22.0软件进行分析,计数资料采用频数表示,组间比较采用Fisher确切概率法。Spearman秩相关分析判断疾病的严重程度(失盐型、单纯男性化型)与基因型分组的关系。以P≤0.05表示差异具有统计学意义。
2 结 果
2.1 CYP21A2基因型64例患儿中,76.6%为复合杂合突变,23.4%为纯合突变。11例患儿存在3处突变。临床分型中,失盐型40例(62.5%),单纯男性化型24例(37.5%),见表1。
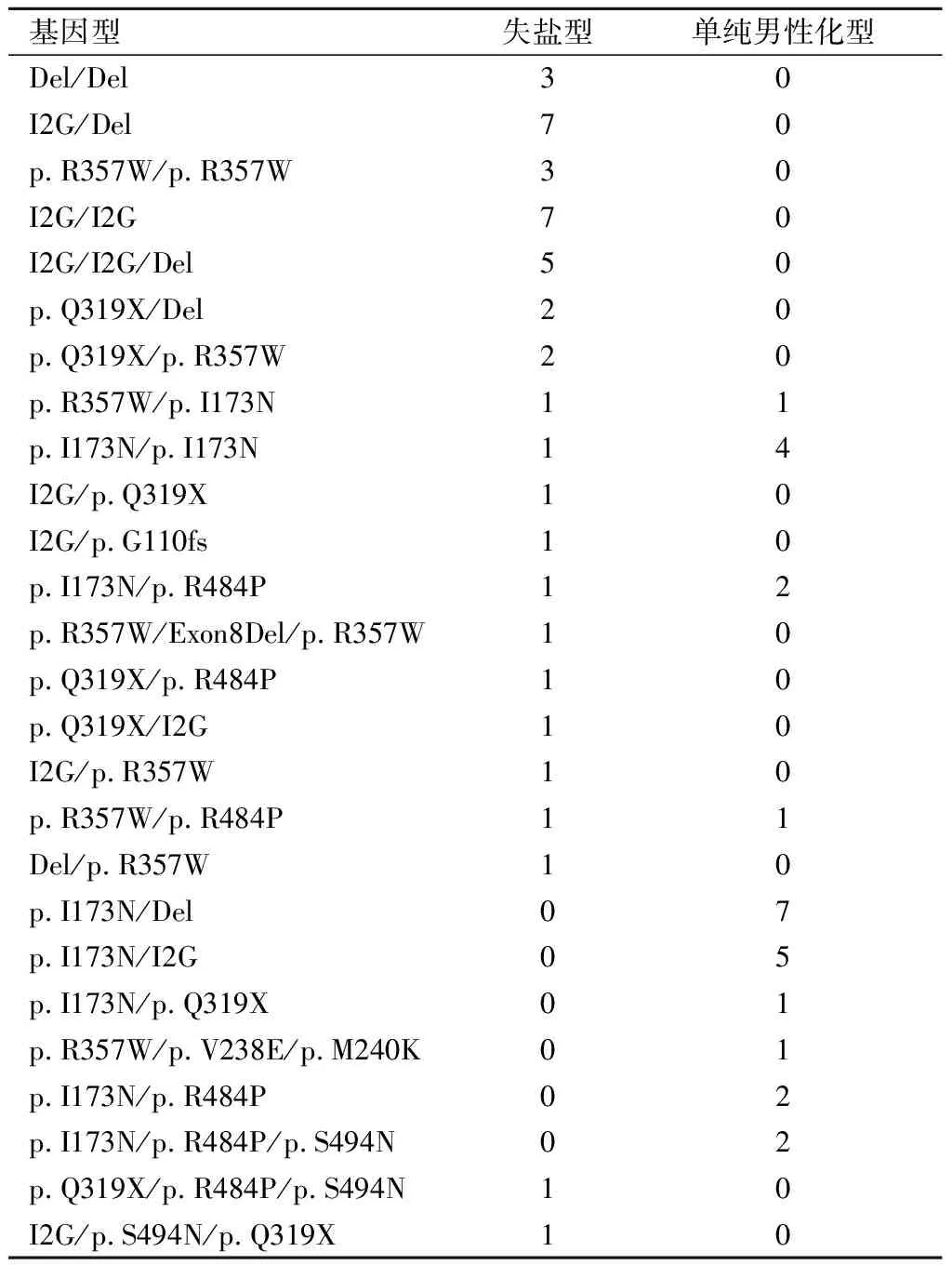
表 1 经典型21-OHD患儿基因检测结果(n)
2.2突变频率分布64例患儿共检出139处突变,包括10种不同的突变,无新发突变位点检出。总体突变频率较高的为I2G、p.I173N、Del等。其中大片段缺失占20.9%,微转换占77.7%。失盐型、单纯男性化型基因突变频率差异有统计学意义(P<0.05)。失盐型最常见的突变位点为I2G(39.8%)、Del(25.0%)、p.R357W(15.9%)、p.Q319X(8.0%)。单纯男性化型最常见的突变位点为p.I173N(51.0%)、Del(13.7%)、I2G(9.8%)、p.R484P(9.8%)。见表2。

表 2 经典型21-OHD患儿总体突变频率的分布情况
2.3基因型与临床分型关系总体基因型预测临床分型准确率为91.8%。极重度突变组对失盐型的阳性预测率为86.7%(13/15),重度突变组对失盐型的阳性预测率为100%(22/22),中度突变组对单纯男性化型的阳性预测率为 87.0%(20/23)。极重度突变组、重度突变组、中度突变组基因型与经典型21-OHD临床分型存在显著正相关(rs=0.691,P=0.00)。说明基因型可以很好地预测21-OHD临床分型。基因型为极重度和重度突变组的患儿更倾向于较严重的失盐型,基因型为中度突变组的患儿倾向于严重程度较轻的单纯男性化型。结果显示,单纯男性化型患儿占比随基因突变对应的残留21-OHD活性增加而逐渐增多。见图1。
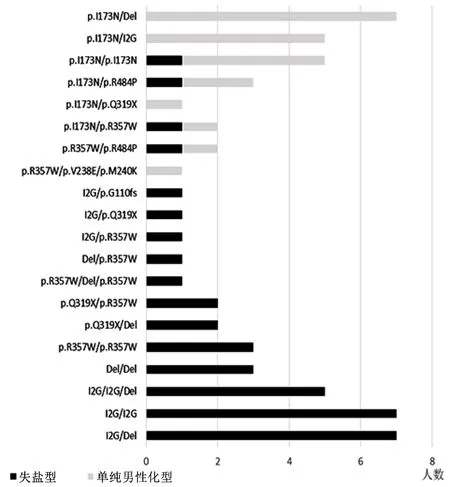
图 1 经典型21-OHD患儿基因型-表型关系
3 讨 论
CYP21A2基因位于6号染色体短臂(6p21.31),在重组活性很高的主要组织相容性复合体III区域内[11]。该区域组成复杂,基因在大小和拷贝数上存在很大变异[12-13]。由于CYP21A2基因结构的特殊性,推荐先使用MLPA检测基因缺失,再通过二代测序检测点突变[14-16]。本研究共检出10种不同的突变,最常见的3种为I2G、p.I173N、Del。
目前报道与21-OHD有关的突变有1300多种,其中有230种会影响人类健康[6]。230种致病突变中156种会导致经典型,74种会导致NC 21-OHD[6]。其中有10种突变最为常见,可以帮助解决大多数临床问题。包括大片段的基因缺失和转换、 8种CYP21A2与CYP21A2P来源的点突变(p.P30L、I2G、p.I173N、E6 cluster、p.V281L、F306+T、p.Q318X、p.R357W)、外显子3的p.G110fs(8个碱基对丢失)。这10种突变占所有突变的绝大多数,本组91.3%的基因突变在此范围内。本研究患儿失盐型占62.5%;单纯男性化型占37.5%,与文献报道[17-18]基本一致。
本研究湖北地区64例患儿中,21-OHD儿童的基因型-表型一致性高,重度突变组患者的临床表型与预期表型的一致性达100%,高于其他大型研究中报告的阳性预测值76%~96%[4,18-19]。但极重度突变组有2例表现为单纯男性化型,基因型分别为p.R357W/p.R484P、p.R357W/p.V238E/p.M240K。p.R357W理论上与失盐型相关。动物细胞实验证明,p.R357W突变会严重破坏21-羟化酶的活性[20]。p.R484P理论上也与失盐型相关。外显子10上两个GG碱基被C碱基对取代,终止密码子提前出现,导致21-羟化酶完全失活。本研究4例中仅1例表现为单纯男性化型,可能与部分患者产生少量具有活性的21-羟化酶有关[19]。外显子6的Cluster 6E三种错义突变(p.I236N、p.V238E、p.M240K)常同时存在。这种2种或3种突变的串联出现会使21-羟化酶失去G螺旋结构,干扰了酶与底物的结合,使21-羟化酶完全不能发挥其应有的催化效应。p.I236N、p.V238E、 p.M240K的单独突变对21-羟化酶活性不会产生大的影响[21]。中度突变组预测表型为单纯男性化型,但有3例表现为失盐型,基因型分别为p.I173N/p.I173N、p.I173N/p.R484P、p.R357W/p.I173N。可能与21-羟化酶在转录、调控、翻译中的细微变化有关[21]。本研究4例未知突变组均携带p.S494N突变,2例失盐型和2例单纯男性化型。该变异的临床意义尚不明确[22]。有研究曾报道1例p.S494N纯合突变的女性患者诊断为失盐型[23]。
近年来,21-OHD的诊疗模式逐渐变化。21-OHD的新生儿筛查在我国各地区逐渐普及。新生儿筛查可检出17-羟孕酮升高的患者。但仅通过激素检测不能预知患儿的疾病的严重程度、确定具体的临床分型[24]。较严重的失盐型与症状较轻的单纯男性化型用药的种类和剂量不同。本研究建立湖北地区21-OHD的基因型与临床表型的对应关系,可为临床医生提供基因型-表型框架。患儿的基因分组为极重度或重度突变组时,主要考虑失盐型;中度突变组需主要考虑单纯男性化型。基因型与临床表型的特定关系可直接服务于临床诊断,为新生儿筛查阳性患儿临床分型提供依据,以实现更精准的治疗。但基因型预测也存在少量不确定因素,需要结合临床体征、生化检测以及诊疗过程的密切随访来弥补。
21-OHD基因型-临床表型对应关系,可指导氢化可的松用药剂量、缩短到达维持剂量需要的时间、降低对患儿成年后身高的影响、尽可能避免外周性性早熟和雄激素过多综合征的发生。女性外生殖器男性化的患儿也可及时找出病因[25]并及时治疗。基因型预测为失盐型的患儿,推荐使用氢化可的松联合氟氢可的松。这种精准的治疗方案可最大程度避免肾上腺危象、减少高钾低钠血症发生、减少住院次数、缩短住院时间。基因型预测为单纯男性化型的患儿,可避免使用过量的氟氢可的松。降低单纯男性化型患儿发生医源性高血压风险。
综上,湖北地区儿童经典型21-OHD基因型预测疾病临床表型可靠性高。患者基因型为极重度或重度突变组时,主要考虑失盐型;基因型为中度突变组时,主要考虑单纯男性化型。本研究21-OHD基因型-表型对应关系,为湖北地区新生儿筛查阳性患儿提供了诊断和治疗依据。
