Ir0.89Ni0.11水合氧化物纳米颗粒电催化水氧化机理研究
2020-11-25田千红刘淑玲
王 超, 田千红, 柴 乐, 刘淑玲
(陕西科技大学 化学与化工学院, 陕西 西安 710021)
0 引言
由于风能和太阳能等可再生能源的不连续性,人类亟需大规模储存能量的方法,如通过水分解成氢气或二氧化碳还原来储存多余的能量[1-3].将水还原为氢气和二氧化碳还原为低碳燃料都发生在设备的阴极,同时,在阳极需要电子供体.因而,阳极的电催化氧化水是最有前景的方法[4].
水氧化反应是一个复杂的过程,涉及4H+和4e-的转移以及O=O的形成,需要极高过电位[5].基于铱的催化剂是目前唯一在酸性条件下具有良好催化活性和稳定性的催化剂[6,7].在氧化铱晶格中掺杂第二种过渡金属(Ni[8,9],Cu[10,11],Co[12]等)能有效提高催化活性、降低成本.其中,Ni作为一种廉价的掺杂剂可明显提高铱基催化剂的电催化水氧化活性[13-16],但其活性提升机理仍有争论.Shuai等[17]发现掺杂Ni提高了IrOx材料的水氧化活性以及导电性,但并不改变反应途径.Moghaddam等[8]和Hong[18]等观察到水氧化活性的增强并不仅仅是由于表面电化学活性位点数目的增加而引起的,这引发了对改善水氧化活性的结构相关因素的研究.Tobias等[13]利用热制备的Ir-Ni氧化物膜,将反应表面-OH基团的覆盖率与水氧化活性相联系,在实验中,12 at%的Ni可以稳定在混合氧化物基体中,并有助于提高水氧化活性.目前发表的机理研究都是基于热处理后的大块Ir-Ni氧化物膜,没有关于Ir-Ni水合氧化物纳米颗粒电催化水氧化的报道.由于不同的合成方法都会对纳米材料的催化活性产生很大的影响,因此对Ir-Ni水合氧化物纳米颗粒的机理研究将有助于理解掺杂Ni对IrOx电催化水氧化活性的影响.
本实验采用了电化学方法,在接近实际质子交换膜水电解槽的工作条件下,使用一种简单的水溶液制备方法对比研究Ir和Ir0.89Ni0.11水合氧化物纳米颗粒电催化水氧化的动力学过程[8].
1 实验部分
1.1 原料及药品
高氯酸,硫酸,氢氧化钾,磷酸,高氯酸钠,碳纤维纸,三水合氯化铱,氯化镍,叔丁醇,Nafion(5 wt%)等.
1.2 水合氧化物纳米颗粒的制备
在20 mL含有0.070 6 g IrCl3·3H2O(0.2 mmol)和0.003 2 g(0.025 mmol)NiCl2的溶液中加入3.1 mL 0.8 M KOH.加入KOH溶液后,在空气中搅拌3天,得到Ir0.89Ni0.11HO-np.制备Ir HO-np过程中不添加NiCl2,且加入2.0 mL 0.8 M KOH.Ir1-xNixHO-np悬浮液用含有适量Nafion的水稀释5倍,最终得到1 wt% Nafion/(Ir+Ni的总质量)的悬浮液.将10μL的Ir HO-np/Nafion悬浮液滴在抛光的玻碳电极(表面积0.196 cm2)上(载量3.4μg Ir)[8].
制备获得的纳米颗粒加入叔丁醇分离,干燥后制得Ir0.89Ni0.11水合氧化物纳米颗粒.
1.3 表征
Ir0.89Ni0.11水合氧化物纳米颗粒直接用于XRD(X′Pert Pro MPD),XPS(Kratos Axis 165)和HRTEM(Titan 80-300 LB)分析.
通过滴加1 M KOH 到0.1 M H3PO4溶液制备不同pH值的溶液.在Tafel斜率测量中,采用x M HClO4+(1-x)M NaClO4来保持离子强度恒定.参比电极为饱和甘汞电极,石墨棒为对电极.将工作电极的电势保持30秒得到稳态Tafel斜率.根据式(1)计算过电位(η):
η=ESCE+0.059pH-1.23
(1)
在变温研究中,以自制的可逆氢电极为参比电极,溶液0.1 M HClO4.根据测量的电势(ERHE)和式(2)计算不同温度下的过电位:
(2)
式(2)中:n=4,F是法拉第常数,假设ΔS和ΔH与温度无关,取CRC公布的热力学常数中的值.
利用Origin 9.0 Pro软件进行三维回归.对于动力学同位素效应研究,在D2O中制备了0.1 M DClO4,参比电极为自制可逆氘电极.根据式(3)将测量的电势(ERDE)转化为在D2O中的水氧化的过电位:
(3)
式(3)中:ΔS和ΔH为该反应标准熵变和标准焓变.

(4)
式(4)中:ΔH= 294.56 kJ mol-1和ΔS=171.44 J mol-1K-1.用交流阻抗测量并进行iR补偿.
2 结果与讨论
2.1 水合氧化物形貌和晶相分析
图1(a)说明Ir0.89Ni0.11HO-np平均粒径为1.33 nm,与相同方法制备的Ir HO-np的晶体粒径分布相似[8].Ir0.89Ni0.11HO-np的面间距分别为0.22和0.25 nm,对应于IrO2四方结构的(200)和(101)面.图1(b)表明Ir和Ir0.89Ni0.11HO-np与具有P42/mnm (JCPDS No.86-0330)空间群的IrO2有相似的结构[19].Ir HO-np在2θ= 28 °和40 °有两个小峰表明结晶度相对较好,而Ir0.89Ni0.11HO-np中的宽峰表明结晶度低,这可能是Ni掺入IrO2晶格引起结晶度变差所致.

(a)Ir0.89Ni0.11 HO-np的的高分辨透射电子能谱

(b)Ir0.89Ni0.11 HO-np和Ir HO-np的X射线衍射图图1 高分辨透射电子能谱以及X射线衍射图
2.2 XPS
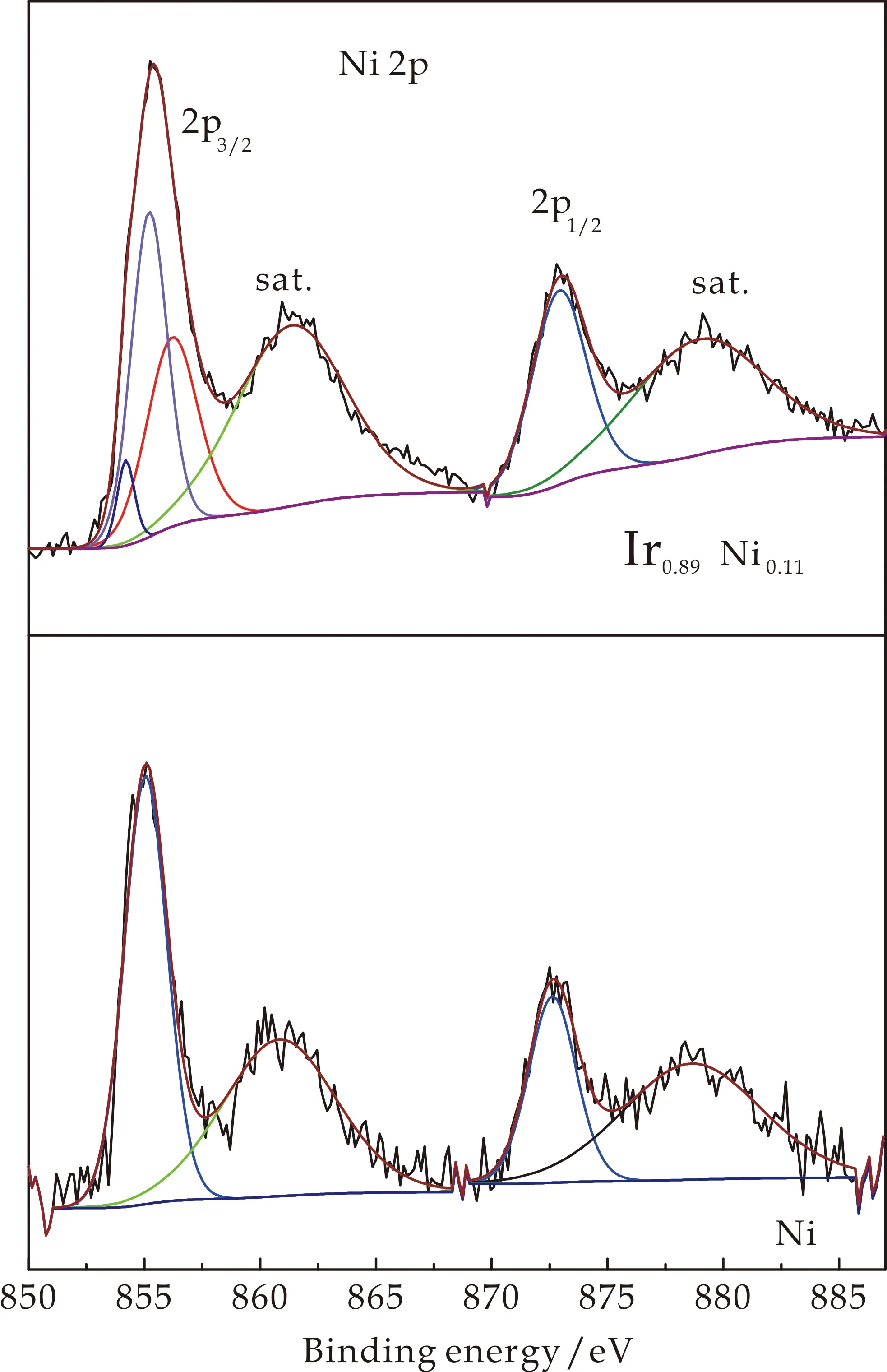
图2为Ir、Ir0.89Ni0.11和Ni HO-np的XPS谱图.图2(a)中,Ir0.89Ni0.11HO-np中的4f7/2(61.6 eV)和4f5/2(64.5 eV) Ir的结合能以及Ir HO-np中的4f7/2(61.9 eV)和4f5/2(65.0 eV)峰位置表明Ir在两者中是4+氧化态[20].4f峰向低结合能方向移动表明Ir0.89Ni0.11HO-np中Ir位的电子密度较高.在Ir0.89Cu0.11HO-np中的Ir 4f结合能中观察到类似的向低结合能方向移动的情形[10].
图2(b)为Ir0.89Ni0.11的Ni 2p区的Ni HO-np的XPS图谱.Ir0.89Ni0.11HO-np (Ni 2p3/2(855.4 eV)和Ni 2p1/2(873.3 eV))的结合能高于Ni HO-np(Ni 2p3/2(854.9 eV)和Ni 2p1/2(872.3 eV))[21].在Ir0.89Ni0.11HO-np的Ni 2p3/2峰可分为853.9 eV(NiO,6%)[22]、855.3 eV (Ni(OH)2,42%)[23,24]和856.3 eV (52%)三个组分.856.3 eV的峰因为其结合能的增加,表明Ni的电子密度降低.Carley等[25]报道Ni3+的结合能为856.1 eV.因此,本实验认为这个峰可能是Ni3+.
图2(c)为Ir、Ir0.89Ni0.11和Ni HO-np的O 1s区域谱图.对于Ir HO-np,分别在530.4 eV、531.3 eV和532.5 eV处出现了三个峰,分别可归属为晶格氧、-OH基团和吸附的H2O[13,22].对于Ni HO-np,只在531.0 eV和532.3 eV两个分峰,表明Ni HO-np主要成分为氢氧化物.然而,O 1s谱显示Ir0.89Ni0.11HO-np在529.7 eV处有一个新的Ir HO-np没有的峰,可归属为桥联Ir和Ni的氧原子[10,13].
基于HRTEM、XRD和XPS,本文提出Ir0.89Ni0.11HO-np具有与IrO2相似的结构,其中11%的Ir被Ni取代.
为了理解Ir0.89Ni0.11比Ir HO-np电催化水氧化活性高的原因,本实验进行了循环伏安法、水氧化Tafel分析和对[H+]反应级数的研究.

(a)Ir 4f区
(b)Ni 2p区

(c)O 1s区图2 Ir、Ir0.89Ni0.11和Ni HO-np的高分辨XPS谱图
2.3 循环伏安测试
图3(a)、(b)为Ir和Ir0.89Ni0.11HO-np在不同pH磷酸盐缓冲溶液中的循环伏安图.Ir和Ir0.89Ni0.11HO-np在所有的pH条件下都表现出典型铱水合氧化物的可逆的Ir3+/4+和Ir4+/5+峰.Ir3+/4+氧化还原为质子耦合电子转移过程如式(5)所示:
e-+qH+
(5)
通过绘制pH值上的峰值电位可得到对应2.3qRT/F的斜率[26].在图3(c)中,Ir HO-np的Ir3+/4+峰随pH的位移为69 mV,相对应的q=1.2,Ir4+/5+峰随pH的位移为59 mV,相对应的q=1.此位移与Baur等[27]和Gambardella等[28]所述的文献值一致.在图3(d)中,Ir0.89Ni0.11HO-np的Ir3+/4+峰随pH的位移为77 mV,相对应的q=1.3.Ir4+/5+峰随pH的位移为59 mV,相对应的q = 1.1.Ir0.89Ni0.11HO-np的Ir3+/4+和Ir4+/5+峰对[H+]都有较高的依赖性.一般来说,随着中心金属氧化状态的提高,配体水和-OH的酸性增加,pKa降低,氧化过程伴随着解离度的增加.基于此,掺杂Ni提高了Ir HO-np在高氧化态下的酸性.

(a)Ir HO-np在不同pH磷酸盐缓冲溶液中的循环伏安图

(b)Ir0.89Ni0.11 HO-np在不同pH磷酸盐缓冲溶液中的循环伏安图

(c)Ir HO-np在不同pH下Ir3+/4+氧化峰电位(黑方点)和Ir4+/5+氧化峰电位(灰色圆点)图

(d)Ir0.89Ni0.11 HO-np在不同pH下Ir3+/4+氧化峰电位(黑方点)和Ir4+/5+氧化峰电位(灰色圆点)图图3 Ir和Ir0.89Ni0.11 HO-np在不同pH磷酸盐缓冲溶液中的循环伏安图(扫描速率50 mV s-1,N2)
2.4 Tafel分析以及对[H+]反应级数分析
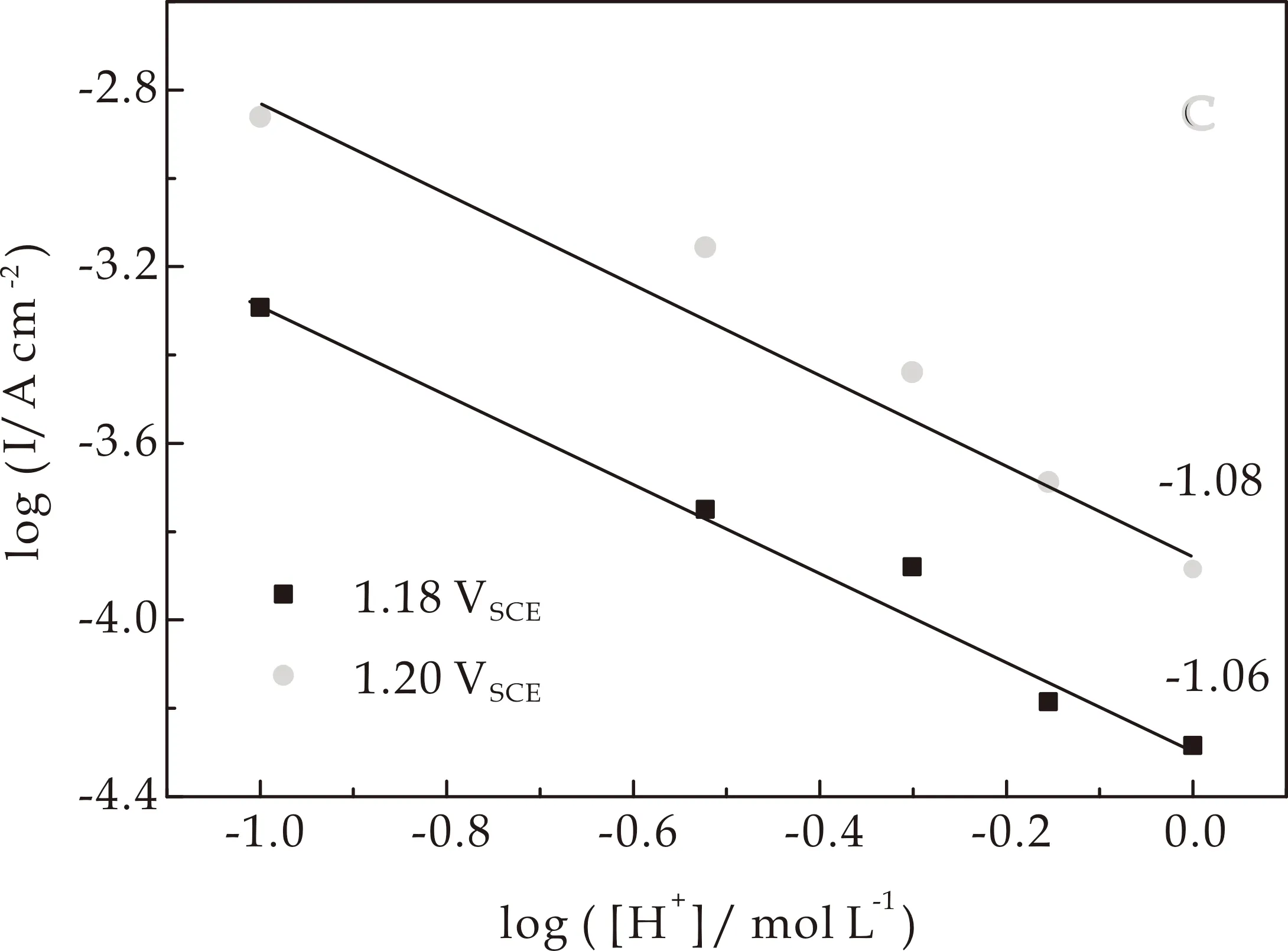
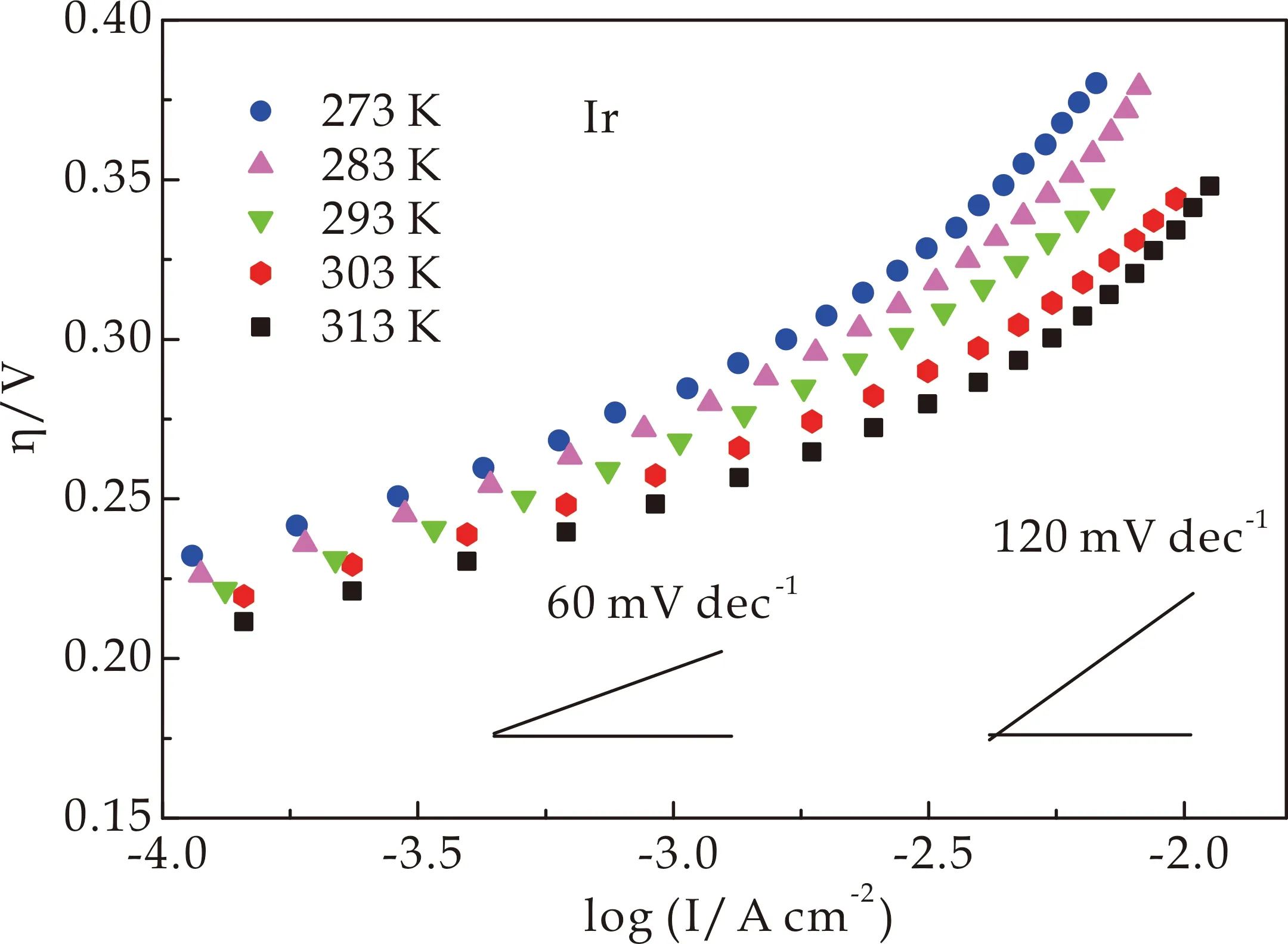
图4(a)为在不同浓度的HClO4溶液中Ir0.89Ni0.11和Ir HO-np的Tafel斜率.在低过电位时,斜率约60 mV dec-1,在高过电位时,约120 mV dec-1,与文献中IrO2值一致[29].图4(b)结果表明,Ir0.89Ni0.11HO-np的Tafel斜率在整个电位范围内为60 mV dec-1,说明水氧化过程决速步是在一个快速电子转移平衡步骤之后的化学步骤.Tafel斜率为120 mV dec-1表明决速步是第一个电子转移步骤(见下文).
图4(c)、(d)为在不同电位下logi对log([H+])作图.斜率表示其对[H+]的反应级数mH+,mH+=(dlogi/dlog([H+]))E.对于Ir HO-np,mH+为-1.08,而Ir0.89Ni0.11HO-np的mH+为-1.18,与之前观察到的Ir3+/4+峰的超能斯特位移一致.且mH+>1表明表面阳离子水合基团参与了水氧化过程[30].

(a)Ir HO-np的Tafel斜率

(b)Ir0.89Ni0.11 HO-np的Tafel斜率
(c)Ir HO-np的水氧化电流对于[H+]反应级数图

(d)Ir0.89Ni0.11 HO-np的水氧化电流对于[H+]反应级数图图4 Ir HO-np和Ir0.89Ni0.11 HO-np的Tafel斜率及在1.18 VSCE和1.20 VSCE下水氧化电流对于[H+]反应级数图
2.5 不同温度下的Tafel分析
图5(a)、(b)为在0.1 M HClO4中,在不同温度下,Ir和Ir0.89Ni0.11HO-np的Tafel斜率.图5(c)、(d)为Ir和Ir0.89Ni0.11HO-np的Tafel斜率的多元正交回归拟合.拟合结果如表1所示.

表1 Ir和Ir0.89Ni0.11 HO-np水氧化参数
Ir HO-np的活化焓为44.76 kJ mol-1.Lettenmeier等[31]报道了IrOx-Ir催化剂的活化焓为40 kJ mol-1.Ferro等[29]发现IrO2-SnO2电极的电催化水氧化活化焓为51.88 kJ mol-1.Ir0.89Ni0.11HO-np的活化焓为35.82 kJ mol-1,表明其活化能垒较低.Ir0.89Ni0.11HO-np的活化焓较Ir HO-np降低8.94 kJ mol-1,这可能与Ir和Ir0.89Ni0.11HO-np表面-OH基团的吸附能以及界面水分子的重组能不同有关.
Kuo等[32]提出,Ir3+/4+的氧化过程可以视为-OH的表面吸附过程.从循环伏安图上看,Ir0.89Ni0.11HO-np的OH吸附峰比Ir HO-np低0.050 V,相当于0.05 eV(4.85 kJ mol-1)的能量差.根据DFT计算,Reier等[4]和Halck等[33]提出表面Ni原子会协同Ru-OH反应中间体转移H,降低过渡态能垒.水分子在界面处的重组也会引起活化焓差异.
因此,本实验认为,在Ir0.89Ni0.11HO-np中,表面的Ir-OH基团会与相邻的Ni-O基团发生相互作用,降低电催化水氧化的活化焓.
(a)Ir HO-np的Tafel斜率

(b)Ir0.89Ni0.11 HO-np的Tafel斜率

(c)Ir的Tafel斜率的多元正交回归拟合

(d)Ir0.89Ni0.11 HO-np的Tafel斜率的多元正交回归拟合图5 不同温度下Ir和Ir0.89Ni0.11 HO-np的Tafel斜率的多维ODR拟合
2.6 动力学同位素效应
图6为0.1 M DClO4中电催化水氧化Tafel斜率.在图6(a)、(b)中,DClO4中的Ir以及Ir0.89Ni0.11HO-np的Tafel行为与在HClO4中相同,说明电催化水氧化机理相同.但在DClO4中的电流密度低于HClO4溶液.在0.28 V过电位下,Ir HO-np动力学同位素效应(KIE)H/D为2.29,而Ir0.89Ni0.11HO-np为2.86.Blakemore等[34]从氢氧化铱溶液中沉积的IrOx在0.1 M KNO3中,1.2 VNHE的动力学同位素效应为2.88.若水氧化过程中包含高价金属离子,次级同位素效应值一般大于1[35].次级同位素效应表明O-H键在决速步中不发生断裂,这与Tafel分析的决速步一致.Ir0.89Ni0.11HO-np上H/D KIE值高,说明相对于反应物,过渡态的O-H键裂解程度较高,这可能是由于表面Ni-O与Ir-OH的相互作用所致.

(a)Ir HO-np的Tafel斜率的比较

(b)Ir0.89Ni0.11 HO-np的Tafel斜率的比较图6 0.1 M HClO4和0.1 M DClO4中Ir和Ir0.89Ni0.11 HO-np的Tafel斜率的比较
2.7 机理讨论
根据以上讨论和实验数据,水氧化的机理如公式(6)~(10)所示:
S+H2O→S-OH+H++e-
(6)
S-OH→S-OH*
(7)
S-OH*→S-O+H++e-
(8)
S-O+H2O→S-OOH+H++e-
(9)
S-OOH→S+O2+H++e-
(10)
式(6)~(10)中:S表示表面活性Ir位点.在Lyon等[36]提出的水合分子模型的基础上,本实验提出Ir和Ir0.89Ni0.11HO-np表面主要由金属为中心含水配位的八面体连接的分散的网状结构组成.
因此,本文提出初始活性位为(-O-)3Ir(OH)(OH2).在Langmuir吸附条件下,假设多相电子转移速率常数服从Butler-Volmer方程,且S-OH表面重组(步骤(7))为决速步,通过以下给出电流密度:
i=4Fk2ΓS-OH
(11)
式(11)中:k2是步骤(7)正向反应的速率常数,ΓS-OH表示S-OH基团的表面覆盖度.将拟稳态近似应用于S-OH得到:

k2ΓS-OH≈0
(12)
式(12)中:β是反应的对称因子.
(13)
式(12)、(13)中:ΓS表示S的表面覆盖度.因此,氧化电流可得:
(14)
在低过电位下,假设k2≪k-1,则方程可简化为:
(15)
式(15)中:k是表观速率常数.因此,Tafel斜率可得:
(16)
相对于[H+]的反应级数:
(17)
这与Ir和Ir0.89Ni0.11HO-np的实验值一致.因此,本文认为在IrOx晶格中掺杂Ni不会改变水氧化的机理.
式(7)是表面吸附S-OH重组为能量不同的表面吸附S-OH*.分子模拟表明,相邻表面吸附S-OH中的氧原子之间的距离小于水中的氧原子之间的距离[37].因此,-OH在表面迁移的速率取决于局部氧环境的重排速率,而非质子传递.表面的初始吸附可以看作是一个快速放电步骤,而后是氢键重组[38].Ni-O基团存在于表面时,Ir-OH与相邻Ni-O基团相互作用生成稳定的表面吸附中间体,从而降低了Ir0.89Ni0.11HO-np的活化焓.图7说明了上述过程.

图7 在决速步之前和决速步的Ir-OH和Ni-O相互作用
从式(17)可得:
(18)
对于高过电位的Ir HO-np,Tafel斜率为120 mV dec-1.Tafel斜率的增加表明决速步变为第一个电子转移的步骤.在高阳极电位下,S-OH表面覆盖度高.在催化剂表面活性较低的区域,H2O的放电速率决定了反应速率.但基于电化学分析和Tafel斜率,本实验无法区分决速步后不同的反应路径.虽然在水氧化过程中,通过傅里叶变换红外光谱[39]和表面增强拉曼光谱[40]可以观察到短暂存在的M-OOH中间体,但仍不清楚氧气的产生是来源于单个Ir位点还是两个Ir位点.
3 结论
此次试验研究了由简单水溶液制备合成的Ir0.89Ni0.11水合氧化物纳米颗粒的电催化水氧化过程,探讨了水氧化活性增强的原因,得出了以下重要结论:
(1)Ir0.89Ni0.11HO-np中Ir-O-Ni基团中的桥联氧原子能从Ni中获得电子,并向Ir位给出电子.因此,改变了-O和-OH在Ir0.89Ni0.11HO-np表面的吸附能,影响其水氧化活性.
(2)Ir0.89Ni0.11HO-np溶液界面内紧密层中的水分子排列更加有序,提高质子传递速率.
(3)Ir和Ir0.89Ni0.11HO-np的水氧化机理相同.界面Ni-O基团与Ir-OH基团上的H原子的相互作用降低了活化焓,因此提升了活性.
(4)次级H/D KIE表示水氧化的决速步不涉及直接O-H键断裂 .
本实验的数据和分析为解释掺杂Ni的Ir HO-np水氧化活性的提高提供了依据.所得结果和用非纳米材料的金属氧化物电极测得的结果一致.本实验强调了通过简单合成二元含水氧化物纳米粒子可以提高活性.这些结果将对工业应用中类似阳极过程的其他混合氧化物体系的设计有所启发.
