头孢克肟颗粒杂质检测的均匀设计及优化
2020-11-24吴建伟余俊先
吴建伟,余俊先
(1.浙江莎普爱思药业股份有限公司研发部,浙江 平湖 314200;2.首都医科大学附属北京友谊医院药学部,北京 100050)
头孢克肟为第3代口服头孢菌素,临床上用于敏感菌引起的呼吸道感染、胆道感染及尿道感染等[1]。《中华人民共和国药典:二部》(2015年版)[2]首次收载了头孢克肟及其制剂,美国药典(40版)[3]、欧洲药典(8.0版)[4]均有收载,欧洲药典提供了有关杂质名称(A、B、C、D、E和F)及其结构式,但未提供分离各已知杂质的色谱参数。本课题组前期研究建立了有关杂质的检测方法,但是在检测影响因素(60 ℃放置30 d,40 ℃放置30 d)样品时,出现了未知杂质,其中一个未知杂质与杂质B分离度无法基线分离,另外系统适应性溶液中已知杂质分离度不能完全达到基线分离,仍有优化空间。本研究采用均匀设计优化了方法,并通过JMP软件[5-8],建立模型,根据模型提出可行的设计空间。
1 材料与方法
1.1 药品与试剂
待测样品:头孢克肟待测样品(纯度98.7%,批号为X180201F,莎普爱思药业股份有限公司)。标准品:头孢克肟对照品(纯度89.0%,批号为KWK1702079,自标),杂质A对照品(纯度98.0%,批号为PIBKW-A,Pharmaceutical Chemistry Laboratory Co.,Ltd,USA),杂质B对照品(纯度92.1%,批号为2757-021A11,TLC Pharmaceutical Standards Ltd,CAN),杂质C对照品(纯度98.0%,批号为PAC-047-10,Aozeal Certified Standards, Inc, USA),杂质D对照品(纯度98%,批号为PITBKW-D,Pharmaceutical chemistry Laboratory Co.,Ltd,USA),杂质E对照品(纯度99.1%,批号为171221-2,Pharmaceutical Chemistry Laboratory Co.,Ltd,USA),杂质F对照品(纯度98.5%,批号为A-02,Pharmaceutical chemistry Laboratory Co.,Ltd,USA)。乙腈(色谱纯,批号为18015097,Tedia Co.,Inc,USA);磷酸二氢钾(分析纯,批号为20170210,北京化工厂);氢氧化钠(分析纯,批号为20170518,北京化工厂);25%四丁基氢氧化铵(TBA,分析纯,批号为20180113,天津光复精细化工研究所)。
1.2 仪器
LC-20AD-PDA型高效液相色谱仪(日本岛津公司);PDA20 A型高效液相色谱仪(日本岛津公司),紫外检测器,光电二极管检测器;AB135-S型十万分之一电子天平(METTLER TOLEDO公司);BSA124S-CW型千分之一电子天平(赛多利斯科学仪器有限公司);FX-312-001/FX-312-002型pH计(雷磁科学仪器有限公司)。
2 方法
2.1 色谱条件及溶液配制
2.1.1 色谱条件:SilGreen色谱柱(250 mm×4.6 mm,5 μm),柱温40 ℃,流速1.0 ml/min,进样体积10 μl,紫外检测波长254 nm。
2.1.2 溶液配制:(1)磷酸盐缓冲液(pH=7.0),取磷酸二氢钾0.68 g,加0.1 mol/L氢氧化钠溶液29.1 ml,用水稀释至100 ml,即得。(2)杂质定位母液,取杂质A—F各适量,精密称定,分别用溶剂溶解并稀释制成每1 ml约含杂质A、B、C、D、E及F各100 μg。(3)杂质定位溶液,精密量取杂质定位母液各1 ml,分别置于10 ml容量瓶中,用溶剂稀释至刻度,摇匀。(4)空白辅料溶液,精密称取空白辅料约950 mg,置于50 ml容量瓶中,加溶剂溶解并稀释至刻度,摇匀,过滤,即得。(5)系统适用性溶液,精密称取对照品约10 mg,精密量取杂质A—F定位溶液各1 ml,置于同一10 ml容量瓶中,加溶剂溶解并稀释至刻度,摇匀,即得。(6)供试品溶液,取40、60 ℃的30 d样品适量,加溶剂溶解并稀释制成每1 ml约含头孢克肟1 mg的溶液,作为供试品溶液。取上述溶液各10 μl,分别注入液相色谱仪。
2.1.3 均匀试验设计:《中华人民共和国药典》收载的头孢克肟有关物质方法色谱柱为C18柱,流动相为TBA溶液(取10%的TBA溶液25 ml,加水1 000 ml,用磷酸调节pH为7.0)-乙腈(V∶V=72∶28)。头孢克肟结构中有2个羧基,根据文献[9],pKa1=2.10(针对羧基)、pKa3=3.73(针对羧基甲氧基亚氨基),pH>其pKa 2个单位时,羧基均解离,在溶液中带负电荷,与TBA形成离子对,头孢克肟及其杂质可能由于羧基的pKa不同,流动相的TBA浓度不同而提供不同的选择性,另外有机相比例也会对各物质保留及分离产生影响。故选择pH、TBA及乙腈比例为考察因素,见表1。

表1 因素及水平表
设计的流动相条件见表2,考察可能对分离度影响较大的3个因素:pH、TBA及乙腈。
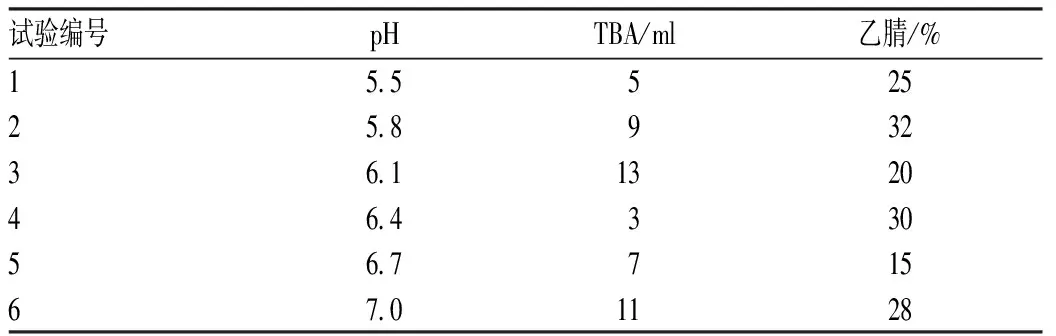
表2 均匀试验设计
3 结果
选择分离度为考察指标,但不同色谱条件下,峰顺序可能发生变化,分离度均为正数,在模型上无法反映出峰顺序颠倒。通常两色谱峰保留时间差>2 min可以较好的分离,暂选择不易分离的关键峰对,其保留时间之差(ΔR)作为考察指标。系统适用性中杂质峰的出峰时间、供试品中与杂质B相邻的未知杂质出峰时间及关键峰对的保留时间之差见表3。

表3 保留时间或保留时间差(min)
主峰及杂质D、杂质B及未知杂质、杂质E及杂质F保留时间接近,不易分析,将这3对作为关键峰对,其保留时间之差分别表示为ΔR(主峰-杂质D)、ΔR(杂质B-未知杂质)、ΔR(杂质F-杂质E)。采用JMP软件以标准最小二乘法回归分析。各回归模型见图1。

A.ΔR(主峰-杂质D)模型参数;B.ΔR(杂质B-未知杂质)模型参数;C.ΔR(杂质F-杂质E)模型参数
可见,ΔR(主峰-杂质D)主要受pH影响,ΔR(杂质B-未知杂质)主要受TBA用量及乙腈交互作用影响,ΔR(杂质F-杂质E)影响因素为各因素主效应及TBA用量与乙腈的交互作用。各影响因素的影响曲面图见图2—4。
pH为影响ΔR(主峰-杂质D)的显著因素,pH在5.5~7时,pH越大,ΔR(主峰-杂质D)越大,见图2。

图2 ΔR(主峰-杂质D)曲面图
pH与TBA交互作用为影响ΔR(杂质B-未知杂质)的显著因素,TBA用量低时,乙腈比例越高;TBA用量高时,乙腈比例越高,ΔR(杂质B-未知杂质)越大,见图3。

图3 ΔR(杂质B-未知杂质)曲面图
pH、TBA、乙腈及TBA、乙腈的交互作用为影响ΔR(杂质F-杂质E)的显著因素,当固定pH=6.5,TBA用量低时,乙腈比例越高;TBA用量高时,乙腈比例越高,ΔR(杂质F-杂质E)越大,见图4。当ΔR(主峰-杂质D)、ΔR(杂质B-未知杂质)、ΔR(杂质F-杂质E)均>2 min时,等高线图见图5—7。

图4 ΔR(杂质F-杂质E)曲面图
TBA为13 ml时,在白色区域中[模型预测ΔR(主峰-杂质D)、ΔR(杂质B-未知杂质)、ΔR(杂质F-杂质E)均>2 min]可选择pH=6.5、乙腈23%为中点,设计空间为pH=6.5±0.25;乙腈为23%±2%,见图5。

图5 TBA 13 ml等高线图
TBA为10 ml时,在白色区域中[模型预测ΔR(主峰-杂质D)、ΔR(杂质B-未知杂质)、ΔR(杂质F-杂质E)均>2 min]可选择pH=6.5、乙腈23%为中点,设计空间为pH=6.5±0.25;乙腈为23%±2%,见图6。

图6 TBA 10 ml等高线图
TBA为8 ml时,模型预测ΔR(主峰-杂质D)、ΔR(杂质B-未知杂质)、ΔR(杂质F-杂质E)均>2 min的区域已不存在。综上,流动相可行的设计空间之一为pH=6.5±0.25,乙腈为23%±2%,TBA为10~13 ml,见图7。采用pH=6.5,乙腈23%,TBA 13 ml,分离度较好,见图8—9。

图7 TBA 8 ml等高线图

图8 优化后系统适用性图

上曲线为60 ℃、30 d样品;下曲线为40 ℃、30 d样品
4 讨论
在新药研究领域,工艺优化、处方筛选的过程中需要对多影响因素及水平进行比较,考察其对结果的影响并优化。目前的相关研究多采用正交设计、均匀设计和效应面法对条件进行优化。正交设计是利用一套系统的正交表将多因素和多水平进行组合均匀搭配,合理安排,从而减少实验次数,同时提供较多的信息[10]。均匀设计,又称均匀设计试验法,或空间填充设计,是基于数论方法推导出来的一种实验设计方法[11-15]。效应面法是利用实验设计并通过实验得到一定数据,采用多元二次方程来拟合因素和效应值之间的函数关系,通过对回归方程的分析来寻求最优工艺参数,解决多变量问题的一种统计方法[16-17]。
从整体评价来看,效应面法要优于均匀设计法和正交设计法,均匀设计法优于正交设计法,效应面法在多因素、多水平的试验方面比均匀设计法更全面,比正交试验法更简化。均匀设计与正交设计相比,更适用于多因素多水平的试验,进一步增强正交设计实验点在空间具有“均匀分散性”的优点,与效应面法相比对一般效应值出现在最佳实验条件区域附近的变化较为灵敏。根据均匀设计法在这一方面的突出特性,本研究将均匀设计应用到头孢克肟颗粒的杂质检测,得到了方法可行的设计空间。本研究结果表明,采用均匀设计和JMP模型处理可以快速优化头孢克肟颗粒有关物质检测的方法,且优化后的方法快捷有效。
