N-烷基乙二胺-双(三氟甲基磺酰)亚胺型质子化离子液体的分子间氢键作用
2020-11-20张欣辰徐宇花儿
张欣辰,徐宇,花儿
(北方民族大学化学与化学工程学院/国家民委化工技术基础重点实验室,宁夏 银川 750021)
离子液体(ionic liquids,ILs)是指由有机阳离子和无机或有机阴离子构成的、熔点在100 ℃以下的熔融盐类[1-3],具有不易挥发、熔点低、液态范围宽、良好的离子传导性及较高的热稳定性等独特的物理化学性质[4-7],因此,ILs离子液体在催化化学、材料科学、电化学、分离技术等不同研究及应用领域受到广泛的关注,并在有机合成、温室气体的吸收、过渡金属离子的萃取及溶解等诸多领域也具有广泛的应用前景[8-9]。
离子液体一般分为非质子型离子液体(aprotic ionic liquids,AILs)和质子型离子液体(protic ionic liquids,PILs)。PILs是由Brønsted酸与Brønsted碱通过酸碱中和一步反应所形成的一类具有特殊性质的离子液体[10],是离子液体的一大分支。PILs除了具有典型ILs的性质以外,因其结构中具有能够迁移的质子而构建氢键网络,表现出较强的极性,扩展了其应用范围,例如,在燃料电池的研究领域,利用其较强的极性作为过渡金属离子的萃取剂及溶解剂,在金属纳米材料领域更加受关注[11-12]。
本文研究[HAlkyl][TFSA]是由Brønsted酸双(三氟甲基磺酰)亚胺(HTFSA)的质子转移至Brønsted碱N-烷基(己基、辛基、异辛基)乙二胺形成的3种PILs,其化学结构式见文献[4]。[HAlkyl]+中氢键供体(如烷基乙二胺中的N—H)与TFSA-中的具有孤对电子的氢键受体间形成N—H…N或N—H…O型氢键,而氢键的强弱对PILs的物理化学性质具有较大的影响[13]。因此,本文研究通过M06-2X方法,在6-311G(d,p)基组条件下计算分析[HAlkyl][TFSA]分子间氢键相互作用,为了预测[HAlkyl][TFSA]型PILs的物理化学性质及PILs的应用研究提供理论基础。
1 计算方法
密度泛函理论(density functional theory,DFT)中的M06-2X方法在描述氢键作用时具有其独特的优点,该方法考虑了色散作用的影响,因此,可以很好地满足对计算的准确性和时效性的要求[14]。本研究选用M06-2X方法,在6-311G(d,p)基组的条件下对[HAlkyl][TFSA]型3种PILs进行几何构型优化、红外光谱分析、电荷分布分析、二阶微扰能的计算,并用分子中原子(atoms in molecular,AIM)理论计算其主要氢键部位的电子密度、拉普拉斯值。本文研究中所有计算工作利用Gaussian 09[15]和AIM2000[16]程序完成。
1.1 相互作用能的计算
选用M06-2X方法及6-311G(d,p)基组,先对[HAlkyl]+[TFSA]-(Alkyl=Hex,Oct,EtHex)阴阳离子对的几何构型进行优化,计算其分子振动频率,计算中确保所优化的构型为势能面上的局部极小点,且均没有虚频;再比较[HAlkyl]+[TFSA]-阴阳离子间的相互作用能(ΔEint),获得[HAlkyl]+[TFSA]-离子对的最稳定构型,其中,ΔEint是指阴阳离子对[HAlkyl]+[TFSA]-的电子能量(Eion pair)与阳离子[HAlkyl]+(Ecation)和阴离子[TFSA]-(Eanion)能量的差[17],相对相互作用能的计算公式如下:
ΔEint=Eionpair- (Ecation+Eanion),
(1)
(2)
ΔEint(电子能)计算中进行零点振动能(zero point energy,ZPE)校正,并利用Boys和Bernardi方法考虑基组重叠误差(basis set superposition error,BSSE)[18],最终以ΔEintBSSE作为基准确定不同[HAlkyl][TFSA]型PILs中或每个PIL几种较稳定构型中的最稳定构型。
1.2 NBO分析
采用相同计算方法和基组条件,通过自然键轨道(natural bond orbital,NBO)方法[19]分析[HAlkyl]+阳离子、[TFSA]-阴离子轨道的相互作用及其对阴阳离子间形成氢键稳定化能的贡献,通过二阶微扰理论中二阶微扰能E(2)预测自然轨道对离子对稳定性的贡献。E(2)值的大小反映从氢键供体轨道到氢键受体轨道的电子离域程度及作用的强弱[20],通过E(2)可以推测电子供体-受体相互作用的强度。对于每个供体轨道i和受体轨道j,E(2)与i→j离域作用的关系[20]如下:
(3)
式(3)中,qi是氢键供体轨道占用的电子数,εi和εj表示轨道能量,F(i,j)表示矩阵单元。
1.3 AIM理论分析
基于优化得到的较稳定构型,通过Bader的AIM理论[21]计算氢键部位电子密度ρc值和拉普拉斯值2ρc,探讨[HAlkyl][TFSA]中的成键本质。
2 结果与分析
2.1 构型优化及相互作用能
通过DFT理论中M06-2X/6-311G(d,p)方法及基组条件,首先对2种不同的阳离子[HHex]1+(伯胺质子化)、[HHex]2+(仲胺质子化)及[TFSA]-阴离子3种单体进行构型优化及静电势分析,结果见图1,其中颜色越深,静电势越大(红色到蓝色的电势变化为0.182~-0.182 e)。图1显示:[TFSA]-中N、O原子上的负静电势较大,[HAlkyl]+中氨基氮原子上的正静电势较大;阴离子[TFSA]-结构中N、O原子比F原子更容易与阳离子[HAlkyl]+中氨基上H原子形成氢键。因此,本文研究主要研究[TFSA]-中N、O原子与[HAlkyl]+中的N—H之间的氢键相互作用。
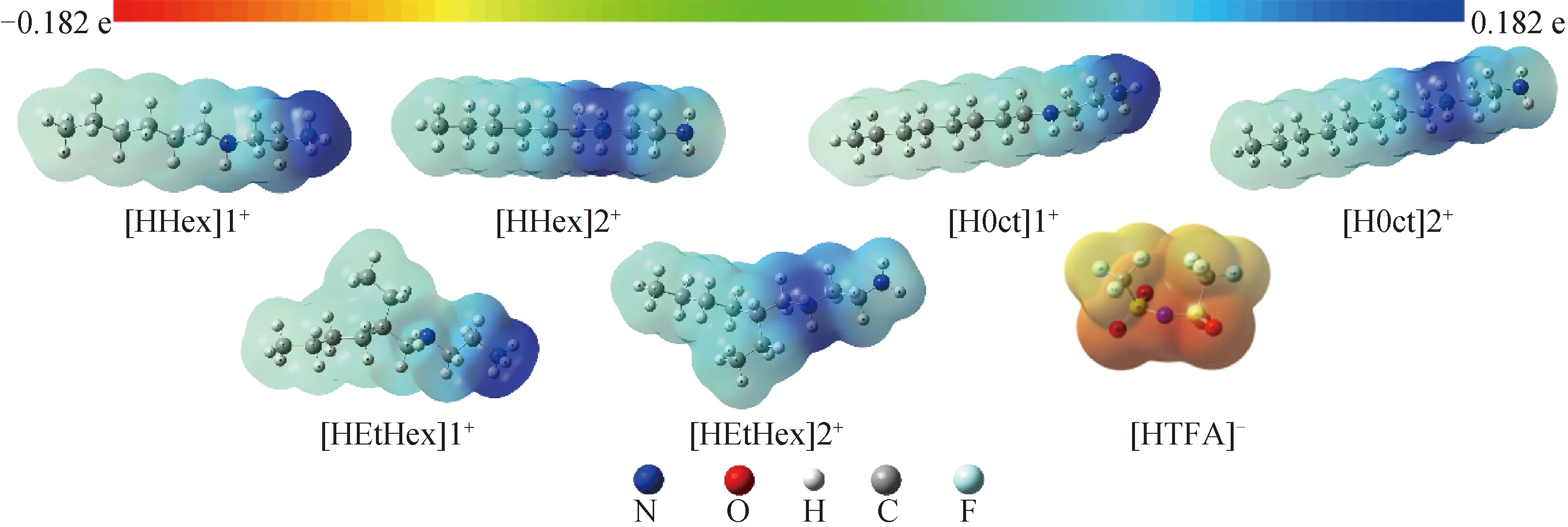
[HHex]1+(伯胺质子化)、[HHex]2+(仲胺质子化);N—蓝色,O—红色,H—白色,C—灰色,F—浅蓝色,S—黄色
设计构型时,考虑可能形成氢键的5个位置(图2),将[TFSA]-排列于所选定[HAlkyl]+周围,分别命名为[HAlkyl][TFSA]S1~S5(S1~S3为伯胺质子化,S4、S5为仲胺质子化)。在M06-2X/6-311G (d,p)水平下,对[HAlkyl][TFSA]S1~S5分别进行构型优化及分子振动频率的计算,结果分别见图2、表2。

图2 [HAlkyl][TFSA]离子对的较稳定构型S1~S5
由表2可以看出:与[HAlkyl]+单体中的N—H键长相比,[HAlkyl][TFSA]中参与氢键的N—H键长的变化最显著,[HAlkyl][TFSA]S1~S5所有较稳定构型中[HAlkyl]+的参与氢键的N—H键长(0.157 8~0.20 20 nm)远大于典型N—H共价键的键长(0.101 nm)。以构型[HHex][TFSA]S1为例,N1-Hp键键长在单体(阳离子[HHex]+)中为0.102 4 nm,而在构型[HHex][TFSA]S1中为0.163 2 nm,键长增加值为0.060 8 nm。从键长的变化值可以初步判断:[HHex][TFSA]S1构型的[TFSA]-中N原子与[HAlkyl]+中的N—H键之间可能形成了N—H…N型氢键。
[HAlkyl]+[TFSA]-阴阳离子对的电子相互作用能(ΔEint)、零点能校正(ZPE)及基组重叠误差(BSSE)校正后的相互作用能ΔEintBSSE见表1,由表1可知:[HAlkyl][TFSA]离子对的ΔEintBSSE值在-88.8~-108.9 kcal/mol范围内。[HAlkyl][TFSA]最稳定构型的ΔEintBSSE(kcal、mol)降低顺序为:[HHex][TFSA](S5=-108.9 kcal/mol)>[HOct][TFSA](S5=-93.9 kcal/mol)≈[HEtHex][TFSA](S1=-94.1kcal·mol-1)。这表明,随着烷基链的增长,阳离子中的电荷分散性增大,[HAlkyl][TFSA]的相互作用能呈现降低的趋势。

表1 ZPE及BSSE校正后[HAlkyl][TFSA]的相互作用能 ΔEint BSSE 单位:kcal/mol
2.2 红外光谱分析
计算M06-2X/6-311G(d,p)水平下较稳定构型[HAlkyl][TFSA]S1~S5(S1至S3伯胺质子化,S4、S5仲胺质子化)的分子振动频率和氢键部位所对应的阳离子单体及形成[HAlkyl]1+[TFSA]-离子对之后的N—H伸缩振动频率值、红移值。结果(表2)表明:
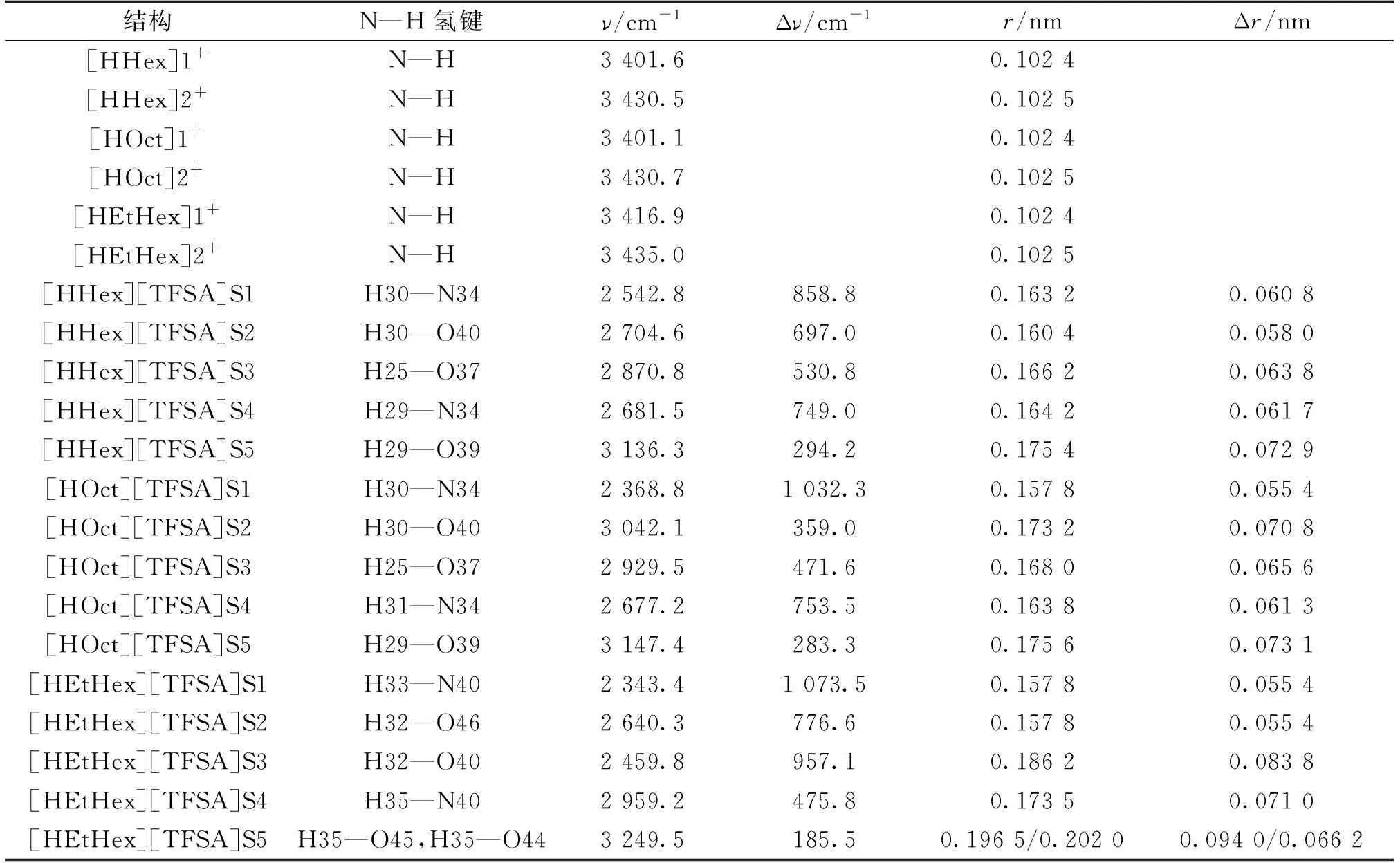
表2 [HAlkyl][TFSA] S1~S5及其阴阳离子结构中N/O—H的键长与振动频率的变化值
(1)[HAlkyl]1+[TFSA]-离子对间主要形成N—H…N或N—H…O型氢键。
(2)所有构型中参与氢键阳离子[HAlkyl]+的N—H键长变化值显著,N—H伸缩振动发生了红移,红移值显著,并且[HAlkyl]1+[TFSA]-的N—H伸缩振动的红移值比[HAlkyl]2+[TFSA]-情况下N—H伸缩振动的红移值较显著,例如,红移值[HHex][TFSA]S1(N—H Δν=858.8 cm-1)>[HHex][TFSA]S4(N—H Δν=749.0 cm-1),[HOct][TFSA]S1(N—H Δν=1 032.3 cm-1)>[HOct][TFSA]S4(N—H Δν=753.5 cm-1),[HEtHex][TFSA]S1(N—H Δν=1 073.5 cm-1)>[HEtHex][TFSA]S4(N—H Δν=475.8 cm-1)。这可能是因为[HAlkyl]+中碳链的增长和支链的引入,使[HAlkyl][TFSA]S1~S5 的分子间(阴阳离子间)氢键相互作用增强,导致N—H伸缩振动频率红移值趋于增大,如红移值[HEtHex][TFSA]S1(N—H Δν=1 073.5 cm-1)>[HOct][TFSA]S1(N—H Δν=1 032.3 cm-1)>[HHex][TFSA]S1(N—H Δν=858.8 cm-1)的。
(3)N—H键键长的变化值与振动频率红移值的变化趋势一致。[HEtHex]1+中N—H键长0.102 4 nm,振动频率3 416.9 cm-1,形成[HEtHex][TFSA]S1型PIL后的N33—H40键长0.157 8 nm,振动频率2 343.4 cm-1,键长及频率变化值分别为Δr=0.055 4 nm、Δν=1 073.5 cm-1。说明[HAlkyl][TFSA]S1构型中形成了N—H…N或N—H…O型氢键,并且N—H…N型氢键相互作用比N—H…O的较强。
2.3 自然布居分析(natural population analysis,NPA)
弥散的电荷分布是离子液体的一个显著的特征,通过电荷分布的分析能够很好解释离子液体的微观结构特征[22]。阴阳离子间的电荷转移对离子液体的稳定性具有重要作用,为了更深入研究[HAlkyl]+[TFSA]-阴阳离子对的电荷分布、电荷转移对分子结构、相互作用能的影响,在M06-2X/6-311G(d,p)水平下对[HAlkyl][TFSA]5种S1~S5较稳定构型进行NPA。表3是电荷变化较为明显的原子的电荷分布值,显示出在氢键相互作用较强部位的原子电荷变化较大,如[HHex]1+中的N28、H30及[TFSA]-中N34、O39的电荷值分别为-0.689、0.444、-1.240、-0.938 e,形成[HHex][TFSA]S1型PIL后分别变为 -0.736、-0.490、-1.323、-0.884e,变化值依次为+0.047、+0.046、+0.083、-0.054 e(+表示电荷增加,-表示电荷减小),其它部位原子上的电荷值没有明显变化。
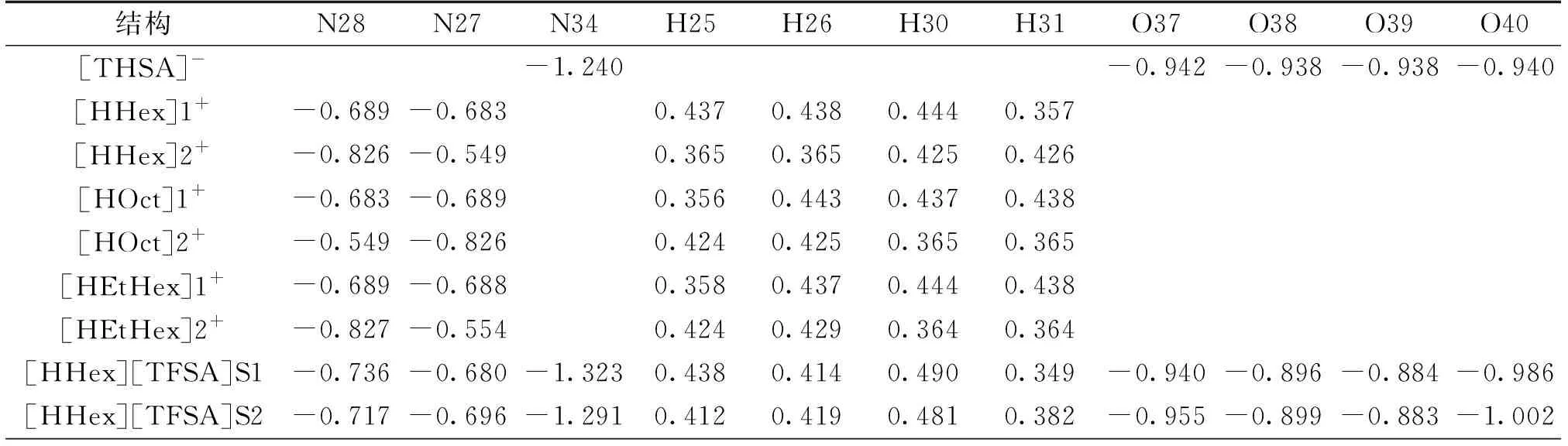
表3 [HAlkyl][TFSA]S1~S5结构中的NPA计算的电荷分布 单位:e
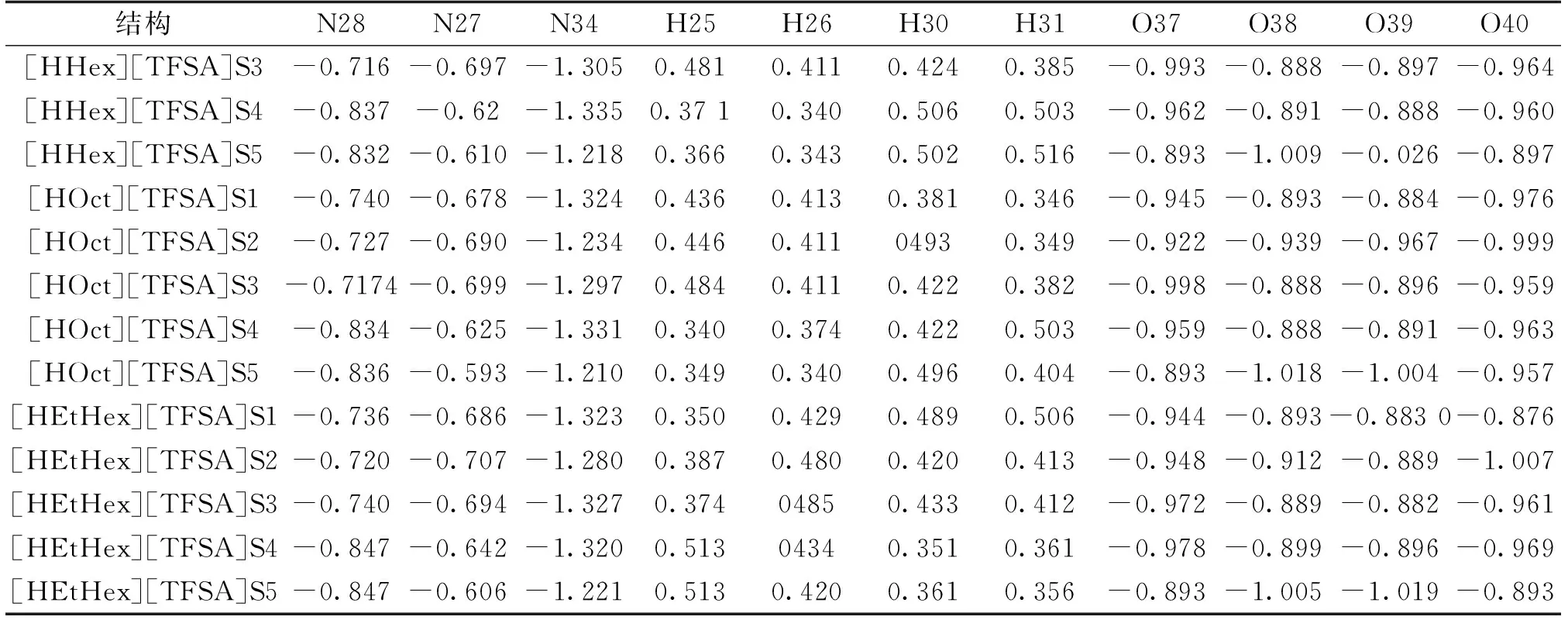
表3(续) 单位:e
另外,根据二阶微扰理论[20]可知,E(2)主要源于[HAlkyl]中N、O原子的孤对电子与[TFSA]中N—H反键轨道之间的相互作用力,即LP (N/O)→σ*(N—H)。[HAlkyl][TFSA]S1至S5的E(2)值见表4。

表4 [HAlkyl][TFSA]氢键部位BCP的二阶微扰能E(2)
从表4可以看出:E(2)值的范围分别为[HHex][TFSA]9.9~53.85 kcal/mol、[HOct][TFSA]23.29~63.51 kcal/mol、[HEtHex][TFSA]9.63~63.66 kcal/mol;LP(N)→BD*(N—H)的E(2)值远大于LP(O)→BD*(N—H)的,表明[HAlkyl][TFSA]中的N—H…N型氢键作用与N—H…O型氢键作用相比,前者中氢键作用较强,例如,[HOct][TFSA]中由大到小依次为63.51(S1,N—H…N)、54.26(S4,N—H…N)、31.19(S3,N—H…O)、24.37(S2,N—H…O)、23.29(S5,N—H…O) kcal/mol;同时,阳离子部位烷基链长的增长和支链的引入对氢键作用强度的影响较大,如[HEtHex][TFSA]的影响与[HOct][TFSA]相近,都大于[HHex][TFSA]。
2.4 AIM分析
AIM理论在氢键作用的研究中应用广泛。AIM以电子密度梯度场中的临界点(critical point,CP)及电子密度分布为基础,定义化学结构、原子及化学键类型。通过对主要氢键部位键临界点(bond critical points,BCPs)的电子密度ρc及拉普拉斯值▽2ρc的分析,能够鉴定阴阳离子间相互作用的成键本质[23]。BCPs的ρc描述化学键的强弱,ρc值越大,化学键能越强,ρc值越小,化学键能越弱;在一定范围内氢键的强弱与ρc的大小成正相关。▽2ρc是ρc的二阶导数,▽2ρc<0表明BCP处的电荷是积聚的,相邻的两原子之间以共价键形式存在,相反,▽2ρc>0表明BCP处的电荷是发散的,相邻的两个原子之间以氢键、离子键等闭壳层作用存在[24]。通常ρc值在共价键中大于0.20 a.u.,在氢键相互作用中小于0.10 a.u.。Hr值作为衡量氢键性质的又一个指标,定义为Hr=Gr+Vr,其中:Hr为哈密顿动能;Gr为拉格朗日动能,是一个与电荷密度ρc、▽2ρc值有关的参数;Vr表征键鞍点处的维里势能。▽2ρc>0、H(r)>0时说明形成了弱氢键,且分子之间的作用以静电作用为主;▽2ρc>0、H(r) < 0时说明形成了中等强度的氢键作用,此时有可能为共价键;▽2ρc>0、H(r)>0 时说明形成了强氢键作用,并且大多数为共价键[25]。
[HAlkyl][TFSA]的主要氢键作用部位BCP处的电子云密度特征见表5,从表5可知:

表5 [HAlkyl][TFSA]的主要氢键作用部位BCP处的电子云密度特征
(1)[HAlky1][TFSA]S1~S5的所有构型中▽2ρc值在0.034 0~0.148 8 a.u.范围内,其值均大于0,并且基本均在典型氢键▽2ρc值(0.020~0.139 a.u.)范围内,同时,其维里势能Vr均小于0,ρc值在0.009 8~0.072 8 a.u.范围内,说明S1~S5构型中均形成了较强的N—H…N或N—H…O型氢键。
(2)[HAlky1][TFSA]S1~S5的ρc值从大到小依次为S1>S4>S2>S5>S3,[HOct][TFSA]的为S1>S4>S3>S2>S5,[HEtHex][TFSA]的为S1>S3>S2>S4>S5。3种PILs构型中,S1的氢键相互作用均最强,且[HHex][TFSA]、[HOct][TFSA]、[HEtHex][TFSA]中S1的ρc值分别为0.064 0 < 0.0728≈0.072 6 a.u.,说明阳离子部位烷基链的增长、支链的引入增强了[HAlky1][TFSA]分子间氢键相互作用。
3 结论
(1)阳离子部位[HAlkyl]+中烷基链的增长或支链的引入,具有减弱[HAlkyl][TFSA]阴阳离子间相互作用能的趋势,3种PILs中[HHex][TFSA]最稳定,即其稳定性大小依次为[HHex][TFSA]>[HOct][TFSA]≈[HEtHex][TFSA]。
(2)[HAlkyl][TFSA]中主要氢键部位BCPs的ρc值在0.009 8~0.072 8 a.u.范围内,均小于0.10 au,▽2ρc值在0.034 0~0.148 8 a.u.范围内,基本均在典型氢键的▽2ρc值(0.020~0.139 a.u.)范围内,进一步说明[HAlkyl]+[TFSA]-阴阳离子间形成了较强的N—H…N或N—H…O型氢键相互作用,[HAlkyl]+阳离子中参与氢键的N—H键长延长,红外谱图显示N—H伸缩振动发生了红移,且红移值显著。
(3)[HAlkyl][TFSA]阴阳离子间因形成了较强的氢键相互作用,使电荷向氢键部位聚集,并提高了[HAlkyl][TFSA]型PILs体系的稳定性,[HAlkyl][TFSA]中N—H…N型氢键作用N—H…O型氢键作用较强。
