脂肪酸代谢与非酒精性脂肪肝疾病关系的研究进展
2020-11-17阎利萍左吉卉吴明江佟海滨
阎利萍 左吉卉 吴明江 佟海滨
温州大学生命与环境科学学院,浙江温州 325000
非酒精性脂肪肝疾病(nonalcoholic fatty liver disease,NAFLD)的发生与肥胖密切相关,在肥胖人群中的患病率达40% ~90%。NAFLD 是一种进展性肝病,从单纯性脂质肝(nonalcoholic simple fatty liver,NAFL)到非酒精性脂肪肝炎(nonalcoholic steatohepatitis,NASH)伴随着持续的坏死性炎症和肝损伤,NASH 患者可发展为进行性肝纤维化,最终发展为肝硬化。目前NAFLD 在全球范围内的患病率急剧上升。NAFLD 发病机制复杂,“二次打击学说”是目前被认可的经典发病机制。脂肪酸进入肝脏后以三酰甘油(TG)的形式在肝实质细胞内大量沉积,胞内代谢逐渐紊乱是疾病发生的第一重打击。当细胞无法将大量游离脂肪酸以TG 的形式储存或超过细胞氧化负荷时,过量脂肪酸产生大量活性氧簇(reactive oxygen species,ROS)引起细胞内质网应激、氧化应激、凋亡、炎症反应等是疾病发生的第二重打击。因此脂肪酸的代谢是NAFLD 发生发展的关键因素,大量游离脂肪酸是引起机体胰岛素抵抗,导致肝细胞脂质沉积以及引起细胞脂毒性的重要原因。了解非酒精性脂肪肝的脂肪酸代谢过程是研究NAFLD 发病机制的基础,同时也为NAFLD 的治疗提供更多潜在靶点。
1 肝细胞脂肪酸来源
1.1 脂肪酸的从头合成
生理条件下,当肝脏内糖原饱和时(约占肝脏重量的5%),肝细胞吸收的额外葡萄糖都被用于合成脂肪酸(图1)。肝脏利用糖类合成脂肪酸这一过程被称为脂肪从头合成(de novo lipogenesis,DNL)。肝脏中参与脂肪从头合成的酶类受固醇调节元件结合蛋白1(sterol-regulatory element binding protein,SREBP1)和碳水化合物反应元件结合蛋白(carbohydrate response element binding protein,ChREBP)的转录调控。乙酰辅酶A 羧化酶(acetyl-CoA carboxylase,ACCs)和脂肪酸合酶(fatty acid synthase,FAS)是DNL 过程中脂肪酸生成的关键酶。肝脏TG 水平由脂肪酸氧化与合成之间的平衡控制,前者受过氧化物酶体增殖激活受体α(peroxisome proliferator-activated receptor α,PPARα)调控,PPARα 可增加脂肪酸氧化、脂解、能量解耦和消耗,后者受SREBF1 调控[1]。在健康肝脏中,DNL不是肝脏脂肪酸的主要来源。但在肥胖和高胰岛素血症情况下,肝脏通过DNL 可产生肝脏脂肪储备25% 以上的脂肪酸[2],而胰岛素抵抗引起的肝脏糖代谢紊乱可能是导致该现象的原因之一。
脂类代谢产物尤其是二酰基甘油(diacylglycerol,DAG)可以抑制肝脏胰岛素敏感性。研究发现NAFLD 患者的DAG 含量比健康个体显著上升[3]。过量DAG 通过激活PKCε 使胰岛素受体酪氨酸残基磷酸化异常,导致PI3K/Akt 信号通路无法激活[4]。转录因子FOXO1 通过对葡萄糖异生酶磷酸烯醇丙酮酸羧激酶和葡萄糖6- 磷酸酶的转录控制,在肝内葡萄糖的合成调控中起关键作用[5]。正常情况下,胰岛素信号激活的Akt 进入细胞核并磷酸化FOXO1,导致其核排斥并泛素化后被蛋白酶体降解,使磷酸烯醇丙酮酸羧激酶转录水平下降,肝内葡萄糖异生率和血糖浓度下降[6]。当肝脏胰岛素抵抗发生时上述过程无法进行,肝内葡萄糖异生率和血糖浓度上升并为DNL 提供了糖环境,解释了在胰岛素抵抗NAFLD 患者中DNL 增加的原因。
1.2 源于外周组织的脂肪酸
脂肪肝所积累的大约60% 脂肪来自于功能失调脂肪组织的脂解[7]。高热量摄入的早期阶段,脂肪细胞积极地储存机体多余的TG,并在禁食期间保持接近正常的脂解速率。随着脂肪组织的显著扩张,脂肪细胞功能障碍导致巨噬细胞趋化因子分泌增多,浸润的巨噬细胞分泌大量的肿瘤坏死因子α(TNFα)导致脂肪组织的慢性炎症状态[8],抑制脂肪细胞TG 的储存,使大量的TG 和游离脂肪酸进入循环系统(图1)。脂肪组织功能障碍可促进胰岛素抵抗相关细胞因子的上调。胰岛素是维持碳水化合物和脂质平衡的必要激素,正常情况下,胰岛素通过抑制激素敏感性脂肪酶(HSL)来抑制脂肪组织中的脂肪分解[9]。在胰岛素抵抗的情况下,胰岛素不能充分抑制HSL,白色脂肪组织的脂解增加,游离脂肪酸释放到循环系统中,造成脂肪在肝脏等器官异位沉积。
1.3 饮食来源的脂肪酸
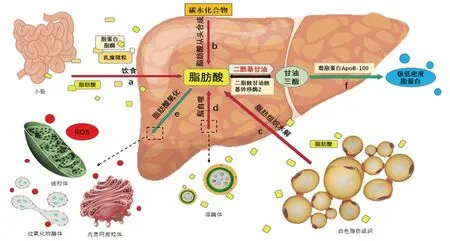
图1 肝脏脂肪酸的代谢途径
食物中的脂肪在小肠上段经胆汁酸盐及脂肪水解酶类乳化水解为甘油、脂肪酸等。中、短链脂肪酸通过门静脉进入到血液循环,长链脂肪酸经小肠上皮黏膜细胞吸收后转化为TG,再与多种载脂蛋白形成乳糜微粒,通过血液循环或淋巴系统运送到身体各组织。进入血液循环的乳糜微粒被脂蛋白脂酶快速分解为脂肪酸、单酰基甘油和乳糜微粒残粒,后者可被肝脏低密度脂蛋白受体和清道夫受体识别并摄取(图1)。长期大量的脂肪被摄取导致血液中游离脂肪酸含量增加,机体利用和储存能力有限,肝脏暴露在一个高水平的游离脂肪酸环境使肝细胞摄入过量脂肪酸,进而导致NAFLD 的发生。
此外,越来越多的证据表明,NAFLD 与果糖的大量摄入相关。肝脏可以快速利用果糖,转化过程不同于葡萄糖,在糖酵解过程中绕过磷酸果糖激酶调节步骤,果糖在果糖激酶和磷酸醛缩酶B 作用下转化为磷酸丙糖和1- 磷酸果糖,此过程既不受胰岛素调节,也不受ATP 和柠檬酸限制,导致肝脏磷酸丙糖含量异常升高。因此,果糖的长期摄入会为肝脏DNL 提供原材料,增加肝细胞中脂肪酸含量。
1.4 源于溶酶体分解代谢的脂肪酸
溶酶体内含多种水解酶可分解脂肪、蛋白质、核酸、多糖等生物大分子。自噬的发生依赖于溶酶体降解系统,是一种对多余或损伤细胞器进行自我消化的过程,为细胞的生长代谢提供原料及能量循环[10]。研究发现,肝细胞内的脂滴可以通过自噬降解,这个过程被称为脂噬[11]。细胞内的脂滴被双膜结构的自噬小体包围并转运到溶酶体后降解成为游离脂肪酸,游离脂肪酸被释放后会被氧化、利用或再次形成TG(图1)。TG 是游离脂肪酸的一种安全储存方式,通过脂噬降解脂滴而释放出游离脂肪酸的代谢过程是否利于改善脂肪肝环境,还需要更多的研究结果去证明,但通过自噬途径吞噬降解损伤的线粒体或内质网在改善NAFLD 中发挥着重要作用。
2 肝细胞脂肪酸的消耗途径
2.1 脂肪酸的氧化
肝脏脂肪酸可以在线粒体、过氧化物酶体以及微粒体中进行氧化分解。线粒体是脂肪酸β 氧化的主要场所,超长链脂肪酸(≥C22)则被转运到过氧化物酶体进行氧化。当细胞内脂肪酸大量存在时,长链脂肪酸可以进行微粒体ω 氧化(图1)。当肝细胞内蓄积大量脂肪酸时,线粒体β 氧化功能超负荷,线粒体内产生大量脂毒性代谢物,从而导致线粒体功能紊乱。同时,过氧化物酶体和内质网对脂肪酸的代偿性氧化增强,而过氧化物酶体和微粒体途径的氧化代谢会产生更多的ROS,造成肝细胞损伤并激活细胞炎症反应。
2.1.1 线粒体β 氧化 线粒体是脂肪酸氧化的主要场所。短、中链脂肪酸可直接透过线粒体膜进入基质进行氧化代谢,长链脂肪酸需经脂酰CoA 合成酶活化生成脂酰CoA,经线粒体内外膜上肉碱棕榈酰转移酶(camitine palmitoyl transterase,CPT)转运进入线粒体基质进行氧化分解[12]。在线粒体内膜的外侧及内侧分别有同工酶CPT-Ⅰ和CPT-Ⅱ,CPT-Ⅰ促进脂酰CoA 转化为脂酰肉碱,然后通过线粒体内膜上的转位酶转运到线粒体内膜内侧,CPT-Ⅱ催化脂酰肉碱释放肉碱后转变为脂酰CoA进入到线粒体基质。脂肪酸经过线粒体β 氧化生成乙酰辅酶A 并进入三羧酸循环。CPT-Ⅰ是脂肪酸线粒体β 氧化的关键酶,当CPT-Ⅰ活性受到抑制或受到转录因子下调时,脂肪酸的氧化会受阻。丙二酰辅酶A 作为脂肪酸从头合成早期的中间体,可抑制CPT-Ⅰ的活性。在脂肪肝中,DNL 十分活跃,在产生大量脂肪酸的同时丙二酰辅酶A 抑制了脂肪酸的β 氧化,导致脂肪酸在肝细胞基质中堆积。PPARα 可上调CPT-Ⅰ的表达,增强脂肪酸在线粒体和过氧化物酶体中的氧化能力,同时增加细胞色素P4502E1 和细胞色素P4504A11 的表达,增强微粒体ω 氧化[13]。
2.1.2 过氧化物酶体β 氧化 过氧化物酶体是细胞内超长链脂肪酸进行氧化的唯一场所,同时也是胆汁酸代谢中间物、多不饱和脂肪酸、长链二羧酸、二十烷类物质等的氧化场所[14]。与线粒体不同,过氧化物酶体不能彻底对脂肪酸进行氧化生成CO2和水,更多的是对脂肪酸进行长度或支链上的修饰。进入过氧化物酶体的脂肪酸经过一定数量的β 氧化循环后,氧化产生的乙酰辅酶A 或丙酰辅酶A,以及被修饰后变短的脂酰辅酶A 将通过肉碱转运机制转入线粒体进行彻底氧化[15]。当肝细胞中蓄积大量脂肪酸时,过氧化物酶体也可以对中长链脂肪酸进行氧化,该过程将产生大量ROS。
2.2 VLDL的输出
肝细胞中脂肪酸以TG 的形式存储,肝脏TG主要以极低密度脂蛋白(VLDL)形式向肝外分泌(图1)。二脂酰甘油酰基转移酶2(DGAT2)催化DAG 共价结合脂酰辅酶A 形成TG,是合成TG 的关键酶。载脂蛋白B-100(ApoB-100)是TG 合成VLDL 过程中重要的前体蛋白。与健康个体相比,杂合失活APOB 突变个体产生的VLDL 更少,肝脏内TG 含量增加3 倍[16]。ApoB-100 定位在内质网膜启动VLDL 囊泡组装,与微粒体TG 转运蛋白(MTP)相互作用促使分泌囊泡成熟。MTP 参与了ApoB-100 在内质网内的转运和转移脂肪到新生脂蛋白颗粒中的过程,是VLDL 合成和分泌过程中的重要蛋白。
正常情况下,肝细胞内脂肪酸和TG 水平升高可促使ApoB-100 表达,肝脏胰岛素水平升高则抑制ApoB-100 表达。在脂肪肝患者中,肝脏脂肪酸含量显著升高并伴随胰岛素抵抗,ApoB-100 表达和VLDL 输出相对增加,但输出速率常常不能代偿肝脏TG 沉积速率。随着蓄积在肝细胞中高浓度的游离脂肪酸引起一系列脂毒性反应,内质网应激的发生导致蛋白质错误折叠,VLDL 合成及运输过程中所需蛋白质合成紊乱,造成TG 在肝细胞中堆积。
3 脂质过氧化
脂质过氧化是氧化应激的一种常见反应,指细胞内ROS 去攻击生物膜的磷脂、酶和膜受体相关的多不饱和脂肪酸,在此过程中可以产生一系列复杂产物和新自由基,即脂质过氧化产物。丙二醛(MDA)、4- 羟基壬烯醛(4-HNE)是两个判断脂质过氧化的醛类重要标志物。4-HNE 是一种对细胞有毒的亲电试剂,含有三个官能团的HNE 活性极强。花生四烯酸、亚油酸和磷脂等游离脂肪酸氧化可以生成大量4-HNE[17],在机体内主要参与4-HNE代谢的酶系统包括谷胱甘肽-S- 转移酶、醛脱氢酶和乙醇脱氢酶等。MDA 是一种酸性双羰基化合物,能与各种生物分子的亲核中心发生多种反应。与ROS 相比,醛基产物具有更强的化学稳定性,可广泛扩散到细胞内外,与亲核性生物大分子如DNA、蛋白质和磷脂等发生反应,当大量脂质过氧化物产生时可使细胞膜的流动性和通透性发生改变,并使细胞结构和功能收到损伤,因此脂质过氧化在NAFLD 的发展中扮演重要角色。
4 脂毒性
脂毒性原指过量游离脂肪酸导致胰岛β 细胞内脂酰辅酶A 增加,导致神经酰胺含量上升并诱导NO 大量产生,引起细胞毒性损伤,现在细胞脂毒性用来广泛描述由脂肪酸及其相关代谢产物造成的一系列细胞损伤。NAFLD 患者的血浆游离脂肪酸含量显著增加是导致机体胰岛素抵抗的重要原因,肝细胞内脂肪酸的超负荷氧化导致线粒体损伤以及ROS 的大量产生,引起氧化应激、内质网应激、炎症反应等,肝细胞内大量脂代谢相关产物如DAG造成肝脏胰岛素抵抗、DNL 增加等,脂肪酸带来的一系列脂毒性效应在NAFLD 的发生发展中起着重要作用,推进了NAFL 向NASH 恶化的进程。
4.1 线粒体损伤与氧化应激
当肝脏中游离脂肪酸及相关代谢产物增加超过细胞代谢负荷能力时,会诱导细胞产生大量ROS,导致氧化应激和线粒体损伤,同时受损线粒体会产生更多的ROS。对健康个体、肥胖的NAFL患者和NASH 患者的肝脏活组织进行高分辨率呼吸测量,发现健康个体与肥胖NAFL 患者线粒体数量无显著差异,但NAFL 患者单个线粒体最大呼吸率是健康个体的4.3 ~5.0 倍;NASH 患者拥有更多线粒体数量,但线粒体最大呼吸率降低31% ~40%;NAFL 患者和NASH 患者的线粒体解偶联和电子渗漏增加[18],表明线粒体超负荷运作和线粒体损伤推进了NAFLD 的发生与发展。除肝细胞对脂肪酸进行氧化分解生成ROS 外,游离脂肪酸也能引起NADPH 氧化酶相关的ROS 产生,以及脂肪酸引起的内质网应激也会刺激更多ROS 的产生[19]。肝过氧化氢酶活性可反映细胞抗氧化防御机制情况,NAFL 患者酶活性没有显著变化,但NASH 患者酶活性明显减弱;DNA 氧化损伤的标志物8- 羟基- 脱氧鸟苷也只在NASH 患者中增加[18]。由此可见,肝细胞内大量ROS 引起的氧化应激促进了NAFL 向NASH 的恶化。
4.2 内质网应激与炎症反应
作为TG 合成和VLDL 组装的场所,内质网在肝脏脂肪酸代谢中扮演十分重要的角色。细胞代谢失衡将导致未折叠或错误折叠蛋白在内质网中聚集,内质网生理功能受损引起内质网应激。大量脂肪酸代谢产生的ROS 引起的氧化系统和抗氧化系统失衡可引起细胞内质网应激,同时脂肪酸可以整合到内质网膜的饱和磷脂双分子层上破坏内质网的形态和功能,引起内质网应激,并激活未折叠蛋白反应(UPR)[20]。UPR 的激活可修复或降解错误折叠蛋白,平衡内质网内环境。UPR 有三个独立的信号转导机制,分别由肌醇依赖性激酶1α(IRE1α)、蛋白激酶R 样内质网激酶(PERK)和活化转录因子6(ATF6)介导[21]。
非应激状态下,IRE1α 与内质网伴侣葡萄糖调节蛋白78(GRP78)结合。在内质网应激状态下,IRE1α 从GRP78 中释放出来并发生低聚,其核糖核酸酶自磷酸化被激活。活化的IRE1α 能催化白介素1β(IL-1β)的基因表达[22],同时催化XBP1mRNA 剪切,具有活性的XBP1 转录因子可诱导下游TNF-α[23],TNF-α 和IL-1β 都是重要的促炎性细胞因子,在脂肪肝炎症发生中起着标志性作用。IRE1α-XBP1s 信号通路在肝脏脂代谢中起着重要的调节作用,该信号通路可通过调节蛋白二硫键异构酶(PDI)和MTP 的表达来调控TG 的转运,为VLDL 在内质网的组装提供底物,但持续过量的TG 将使IRE1α 的核酸内切酶活性受到抑制,使下游脂肪酸氧化和脂质分解相关因子的表达降低,导致肝细胞内脂质的进一步蓄积[24]。
PERK 是位于内质网膜上的跨膜激酶,当应激发生时与GRP78 分离并自磷酸化。白介素23(IL-23)是内质网应激诱导的PERK 下游转录因子C/EBP 同源蛋白的靶基因[25],同时PERK 介导的JAK-1/STAT-3 信号通路的激活可诱导炎症细胞因子白介素6(IL-6)和多种趋化因子的表达[26],促进肝脏炎症的发生。同时在内质网应激时,PERK可破坏C/EBP 的功能,抑制脂代谢相关基因的表达,引起脂肪酸氧化和Apo 分泌过程的紊乱[27]。
ATF6 是一种转录因子,在内质网应激时在高尔基体被蛋白酶S1P 和S2P 剪切重组,活化后的ATF6进入细胞核激活UPR 靶基因,诱导促炎因子的表达。应激引起的ATF6 过表达可激活核因子B(NF-B)介导炎症反应,导致肝脏炎症和受损。不同于其他两条UPR 信号通路,ATF6 的活化可增强PPARα的转录活化,促进脂肪酸氧化,同时上调ApoB-100的表达,促进VLDL 的组装与分泌[28],在NAFLD 中扮演着保护者的角色。UPR 的三个信号转导机制都参与调控肝脏脂代谢过程,与炎症的发生也密切相关。总之,内质网损伤加速了NAFLD 的发展。
5 总结与展望
全球NAFLD 患病率逐年上升,其致病机制复杂并与其他代谢疾病密切相关,作为一种进行性疾病,每一个发展阶段都伴随新的病症,这为NAFLD的预防和治疗带来极大的难度。探究脂肪酸如何诱导了细胞损伤是针对脂代谢过程寻找治疗NAFLD新靶点的基础。目前临床上对该疾病的治疗方案和药物都极其有限,主要以胰岛素增敏和保肝抗炎为主,极少直接针对肝脏脂肪代谢过程。在NAFLD初期,肝细胞内脂肪酸以TG 的形式大量堆积,不伴随炎症反应,因此可通过上调载脂蛋白表达促进VLDL 的外排、注重保护线粒体功能等方式帮助肝脏减少脂肪酸含量,同时通过减少脂类(外源脂肪酸摄取)和碳水化合物类(内源脂肪酸合成)饮食。炎症的发生意味着疾病的恶化,在NASH 时期,过量的脂肪酸已经引起了一系列的肝细胞损伤,在抗炎和胰岛素增敏的同时可干预紊乱的脂代谢过程。脂肪酸不仅是TG 合成的原料,也是引起肝细胞胰岛素抵抗、脂毒性效应、炎症反应的关键,在推进NAFLD 发生发展中起着重要作用。
总之,了解非酒精性脂肪肝中脂肪酸的代谢过程以及引起的细胞损伤,有助于理解NAFLD 的发生和发展机制,为疾病的治疗提供新的作用靶点和干预策略。
