碳自掺杂g-C3N4光催化性能的原位光微量热-荧光光谱研究
2020-11-13马祥英廖艳娟覃方红尹源浩黄在银陈其锋
马祥英,廖艳娟,覃方红,尹源浩,黄在银,陈其锋
(1.广西民族大学化学化工学院,2.海洋与生物技术学院,南宁530008)
石墨相氮化碳(g-C3N4)因具有较好的热化学稳定性、半导体特性和光学特性而受到越来越多的关注,是一种很有前途的无金属二维半导体光催化材料,可催化分解制氢制氧、还原CO2、分解有机污染物[1~3].然而,g-C3N4存在带隙较宽、光生载流子快速重组、比表面积小且表观量子效率较低的缺点,造成其光催化效率较低,对可见光利用有限[4].为了提高g-C3N4的光催化性能,常采用离子掺杂将特定元素引入g-C3N4中取代结构中不同位置的C,N或直接进入g-C3N4层状结构晶格间隙中.晶格结构和电子结构的变化会影响g-C3N4能带结构,同时也会影响g-C3N4光生电子和光生空穴的分离效果及氧化还原能力[5],最终改变g-C3N4的光催化活性.外来元素或离子掺杂在无机半导体光催化剂改性方面应用较多,如用金属(Fe,Pd和Pt等)[6~9]和非金属(S,O和B等)[10~12]来调控g-C3N4能带结构和价带及导带位置.但是,杂离子掺杂也有一些缺点,如光生空穴的氧化能力低、掺杂不对称以及杂质作为光生载流子复合的缺陷[13~15].研究结果表明,自掺杂作为光生载流子的复合中心,可以在不引入杂质和更多缺陷的情况下调整无机氧化物半导体光催化剂的表面性质和电子结构[16,17],以改善其应用性能.Li等[18]以壳聚糖和三聚氰胺为前体,采用超分子自组装的方法构建骨架碳桥g-C3N4,碳桥化的g-C3N4作为一个电子桥,可以有效地将光生电子转移到析氢活性位点,其光催化制氢效率是原始g-C3N4的7倍.Zhao等[19]利用三聚氰胺(CAM)多孔树脂泡沫作为软模板,一步原位合成C自掺杂g-C3N4,研究发现C取代g-C3N4中桥接N原子后,很好调控了g-C3N4样品的微观形貌、电子和能带结构,显著提高了可见光条件下g-C3N4对NO的去除效率.Tong等[20]在蔗糖分子的介导下,利用水热过程将CAM超分子组装成三维微花,然后煅烧后同形转化为由碳涂层g-C3N4纳米薄片组成的g-C3N4@C三维多孔微花,且通过改变糖的种类,发现糖分子的甲酰基团及其自缩聚行为可以控制CAM的组装和随后微花结构的形成,所制备的三维多孔微花g-C3N4@C具有较高的析氧活性.由此可见,g-C3N4的C掺杂可以让C取代g-C3N4中桥接N原子形成骨架自掺杂,也可形成碳涂层沉积在g-C3N4表面上掺杂在晶格间隙中,两种掺杂方式均对g-C3N4的电子导电能力、光生电子-空穴分离效率、光催化性能产生不同影响.但这两种C掺杂分别对g-C3N4的形貌物相结构、能带价态组分和光催化性能产生的影响及其光催化降解机理的不同尚未见报道.
光催化过程涉及因素很多,包括载流电子和空穴产生、迁移、复合等分子和电子层面等微观问题,又涉及表面吸附、表面反应、脱附、扩散等宏观变化,表现为多尺度复杂过程的热力学、动力学问题.光微量热-荧光光谱联用技术能同步获取催化过程的热/动力学和分子光谱信息,能高分辨地体现催化系统中物质本质结构转换信息,并能高精度、高灵敏度地同步在线实时获取整个光催化过程中的特异性光谱和非特异性热谱信息,快速将过程中的热/动力学、光谱动力学及催化过程机理有机地关联,提供了一种耦合热信息和光谱信息认识光驱动过程微观机制的原位联用技术.
单宁酸分子具有多―OH基团,碳含量高,可作为制备碳自掺杂g-C3N4的前体.尿素具有大量的―NH基团,可作为氢键的给体和受体[21].本文首次利用单宁酸和尿素分子之间形成的三维(3D)氢键超分子网络结构,通过调控单宁酸的用量,原位缩聚形成骨架C掺杂和无定形C掺杂的g-C3N4,并对碳自掺杂g-C3N4的物相形貌结构和能带价态组分进行了表征分析,结合紫外-可见吸收光谱和原位光微量热-荧光光谱联用仪获取碳自掺杂g-C3N4降解罗丹明B的原位热/动力学信息和三维荧光光谱信息,探讨了骨架C掺杂和无定形C掺杂的g-C3N4光催化降解罗丹明B的性能和微观机理,为设计和合成高效自掺杂可见光光催化剂提供了新的思路和参考.
1 实验部分
1.1 试 剂
单宁酸、罗丹明B、异丙醇、三乙醇胺和对苯醌,购于上海阿拉丁试剂有限公司;尿素购于上海麦克林生化科技有限公司;无水乙醇购于汕头西陇化工股份有限公司.以上试剂均为分析纯.
1.2 g-C3N4和C掺杂g-C3N4的制备
分别将0,30,75,150,300,525和750 mg的单宁酸加入到15 mL去离子水中,搅拌均匀,分别制得0,2,5,10,20,35和50 mg/mL单宁酸溶液.室温下将15 g尿素置于100 mL加盖坩埚中,分别加入上述单宁酸溶液,搅拌均匀,置于马弗炉内在空气氛围下加热至400℃,恒温2 h,继续加热至550℃并保持2 h,待自然冷却后取出样品,用去离子水洗涤3次,乙醇洗涤3次,于60℃烘干,分别获得g-C3N4和不同掺杂量C改性的g-C3N4光催化剂,分别记为C3N4,C3N4C1,C3N4C2,C3N4C3,C3N4C4,C3N4C5和C3N4C6.
1.3 材料的表征
X射线光电子能谱(XPS)表征采用美国Thermo Fisher Scientific公司的Kα型X射线光电子能谱仪;傅里叶变换红外光谱(FTIR)采用美国Thermo Fisher Scientific公司的MAGNA-1R 550型傅里叶变换红外光谱仪;X射线衍射(XRD)采用德国Bruker-AXS公司的D8 Advance型X射线衍射仪,CuKα辐射,测定范围5°~50°,扫描速率5°/min;热重曲线(TG)采用德国STA449F3公司TG-DSC型同步热分析仪,升温速率10°C/min,测试温度35~800℃;场发射扫描电子显微镜(FESEM)照片采用德国蔡司公司Supra55 Sapphire型场发射扫描电子显微镜;光致发光光谱(PL)表征采用日本日立公司F-4600型荧光分光光度计;紫外-可见吸收光谱(UV-Vis)表征采用美国安捷伦公司Cary60型紫外-可见分光光度计;固体紫外-可见漫反射(UV-Vis DRS)表征采用日本岛津Shimadzu公司UV-2501PC型固体紫外-可见漫反射仪;BET比表面积和孔隙分布采用美国Micromeritics公司TriStarⅡ3flex型全自动比表面及孔隙度分析仪;光催化反应测试采用配有350 W氙灯和420 nm可见滤光片的南京胥江机电厂的XPA-7型光化学反应仪.
1.4 光催化性能测试
在5支20 mL试管中加入10 mL 20 mg/L罗丹明B溶液,再分别加入20 mg不同的光催化剂,超声溶解混合均匀后将其放置在光化学反应仪中,室温下避光以600 r/min转速磁力搅拌1 h,使催化剂和罗丹明B溶液到吸附-脱附平衡,继续搅拌下打开氙灯进行光催化反应,分别在光照0,10,20,30,45和60 min后离心取上层清液,用紫外-可见分光光度计分别测定各时间点的降解率.
1.5 光催化活性物种俘获检测实验
室温下取4支试管,分别加入10 mL 20 mg/L罗丹明B溶液与20 mg光催化剂,超声溶解混合均匀后,避光以600 r/min转速搅拌1 h,使催化剂和溶液达到吸附-脱附平衡,分别加入2.5 mL对苯醌(·俘获剂)、异丙醇(·OH俘获剂)、三乙醇胺(h+俘获剂)和去离子水(空白对比),室温下放置在光化学反应仪中进行光催化反应,在光照30 min后离心取上层清液,用紫外-可见分光光度计分别测定各时间点的降解率.
1.6 光催化降解罗丹明B原位光微量热-荧光光谱表征
将10 mg光催化剂样品和5 mL 20 mg/L罗丹明B溶液置于10 mL试管中并超声混合均匀后,避光以600 r/min转速磁力搅拌1 h.调好参比光源并设置好仪器温度参数,待仪器温度恒定后,取0.5 mL上述混合均匀的溶液置于新型光微量热-荧光光谱联用装置的样品池中,并在参比池中加入0.5 mL去离子水.等待基线走平后,打开光功率为40 W/m2、波长为405 nm的激发光源,进行时长为1 h的光催化反应,获取该温度下光催化剂降解的原位热谱曲线和荧光光谱信息.分别测试各光催化剂在288.15,293.15,298.15和303.15 K温度下的原位光微量热-荧光光谱信息.

Fig.1 XPS spectra of C1s(A)and N1s(B)for the catalysts
2 结果与讨论
2.1 催化剂物相结构和形貌表征
图1的XPS谱图是为了确定C掺杂对g-C3N4化学骨架的影响,催化剂的XPS谱图分析数据见表1.改性后的g-C3N4,其C1s和N1s的XPS谱峰形状与纯g-C3N4样品的基本一致.对催化剂的C1s和N1s进行高斯曲线分峰拟合,C1s可拟合分为284.60,286.24和287.76 eV 3个特征峰,284.60 eV归属于石墨相g-C3N4中含有缺陷sp2杂化C—C键,286.24 eV处的峰归属于未聚合完全的C—NH,287.76 eV处的峰归属于三嗪环结构中sp2杂化的 C—(N)3官能团[22].N1s峰可分为398.61,400.29和401.35 eV 3个特征峰,398.61 eV处的峰归属于sp2杂化的C=N—C官能团中的芳香N(标记为N1),400.29 eV处的峰归属于N—(C)3(标记为N2),401.35 eV处的峰归属于C—N—H化学键[23].由以上N1s和C1s的XPS谱图分析结果,可确认g-C3N4的基本组成单元七嗪杂环(C6N7)存在于各催化剂样品中.随着加入单宁酸浓度的增加,催化剂中C/N比例增大,说明样品自掺杂了C原子,而N1s中N1/N2比例下降,C1s中286.24 eV处的峰面积增大,说明在g-C3N4的骨架中部分N原子被碳原子取代.值得注意的是,从C3N4C4样品开始,其N1s的398.61 eV处和C1s的287.76 eV处的特征峰显著减小,N1s的400.29,401.35 eV和C1s的286.24,284.6 eV处的峰面积显著增加且有显著位移,说明样品中未聚合的C—NH碎片增多,C3N4C4中多余的C可能是以无定形C的形式沉积在样品表面从而导致g-C3N4的聚合度下降.为了进一步证实催化剂样品中多余的C是以通过取代g-C3N4晶格中的N原子,还是以无定形的C沉积吸附在g-C3N4中,将采用FTIR,TGA和XRD等方法对样品做进一步表征.

Table 1 XPS analysis of catalysts

Fig.2 FTIR spectra(A),XRD patterns(B),TGA(C),steady⁃state photoluminescence(PL)spectra(D)of catalysts
催化剂的FTIR,TGA和XRD谱图分别见图2.由图2(A)的FTIR谱图可知,C3N4C1,C3N4C2,C3N4C3样品的FTIR谱线与纯C3N4样品的基本一致,均观察到石墨相g-C3N4的特征吸收峰.700~800 cm-1范围的弱吸收峰归属于碳氮杂环的弯曲振动,810 cm-1处的特征吸收峰归属于三嗪结构的弯曲振动,1200~1600 cm-1范围的特征峰对应典型芳香族C3N4杂环,其中1636和1540 cm-1处的吸收峰对应C=N键,1317和1238 cm-1处的吸收峰对应C—N单键,3000~3700 cm-1范围内的吸收峰对应未缩合的氨基中残留的N—H伸缩振动[24].而C3N4C4,C3N4C5和C3N4C6的FTIR谱图除了具备g-C3N4特征吸收峰,还具有1380 cm-1处的特征吸收峰,可对应石墨碳的伸缩振动,且随着C掺杂量的增大,该特征峰逐渐增强,进一步证实了无定形碳存在于C3N4C4样品中.
由催化剂的XRD谱图[图2(B)]可知,g-C3N4(JCPDS No.87-1526)2个典型的特征衍射峰在g-C3N4和C自掺杂g-C3N4样品均可以检测到,表明C自掺杂不会改变g-C3N4的构相.2θ=27.2°所对应的最强峰归属于(002)晶面,这是一种典型层状堆积的共轭芳香结构,2θ=13.1°的副衍射峰归属于石墨烯材料的(100)晶面[25]. 与纯C3N4样品相比,随着C/N比的增大,C3N4C1,C3N4C2和C3N4C3样品的2θ=27.2°处的特征衍射峰强度增强,说明随着g-C3N4骨架中C的掺杂量增大,样品具有更好的结晶度,更有利于电子转移.而C3N4C4,C3N4C5和C3N4C6样品的(002)晶面峰强度减小,且向低角度偏移至27.1°,说明层间距变大,这是由于C的原子半径大于N,过量C元素的掺杂增大了原子层的堆积间距;2θ=13.1°处的特征衍射峰强度变弱,说明其基本组成单元七嗪杂环(C6N7)有序度下降,不能形成有序的面内重复单元.
由催化剂的TGA谱图[图2(C)]可知,C3N4C1,C3N4C2和C3N4C3与纯g-C3N4的TGA谱图基本一致,在140℃以下轻微的失重峰是由于各样品所吸附的水分挥发所致,除此之外在600℃以下无明显的失重峰,样品均在640℃发生分解,这表明C3N4C1,C3N4C2和C3N4C3样品中没有无定形C存在[18].C3N4C4,C3N4C5和C3N4C6样品也具有140℃以下样品吸附水分挥发所致的失重峰,但在298~420℃范围内出现了失重峰,可归因于无定形碳与空气反应所致,且随着C掺杂量增大,样品失重比例增大.该样品在500℃发生分解,主要是因为无定形C沉积负载在g-C3N4上,晶体有序度下降,相比于纯g-C3N4分解温度下降.通过以上XPS,FTIR,XRD和TGA表征结果的综合分析,充分表明了制备样品时加入少量单宁酸(浓度≤10 mg/mL)时,制备C3N4C1,C3N4C2和C3N4C3时多余的C掺杂进入石墨相g-C3N4骨架并取代其N原子形成骨架碳自掺杂;而单宁酸浓度≥20 mg/mL时,制备C3N4C4,C3N4C5,C3N4C6时多余的C是以无定形形式沉积负载在石墨相g-C3N4上形成无定形C自掺杂.
光激发后产生的光生电子和空穴有2种传输途径:第一种是迁移到光催化剂表面的活性位点处,进行后续的化学反应;另一种是光生电子和空穴直接复合,没有参与进一步的光催化反应.采用光致发光光谱(PL)研究了在395 nm激发光下样品的光生电子和空穴的复合强度[图2(D)].在图2(D)中,未掺杂的纯g-C3N4PL光谱在460 nm处存在一个强的本征荧光发射峰,表明其具有显著电子和空穴复合强度[26].以无定形C自掺杂的C3N4C4,C3N4C5和C3N4C6的PL强度相对C3N4显著下降,但随着C自掺杂量增多,样品的PL强度增加,表明无定形C自掺杂过量不利于电子和空穴分离,这可能是过量无定形C掺杂引起催化剂聚合结构有序度降低从而抑制光生电荷迁移有效性所致.骨架自掺杂的C3N4C1,C3N4C2和C3N4C3相对于无定形C自掺杂样品,其PL强度进一步逐渐减弱,且发光峰出现宽化,这可能是因为形成不均匀局域能级所致,Zhang课题组[27]已经从理论和实验上证实了C自掺杂进g-C3N4骨架后构建的离域大π键大大提高了电子的导电性能,能促进电子和空穴分离.C3N4C3显示将近猝灭的荧光强度,表明其具有良好的光催化特性.
图3(A)为样品在300~800 nm范围内的紫外-可见漫反射吸收光谱.可见,纯g-C3N4的吸收边在470 nm处,这与文献[28]报道的一致.C掺杂后,样品的吸收强度从紫外区域至近红外区域,均有大幅度提高,且光吸收强度增强顺序为C3N4C1>C3N4C2>C3N4C3>C3N4C4>C3N4C5>C3N4C6>C3N4.依据UV-Vis DRS的数据,以纵坐标(αhν)1/2对横坐标Eg作图[图3(B)],曲线直线部分作一条切线使之与横坐标相交,交点即为禁带宽度(Eg).在零电荷点处,g-C3N4的导带电位(ECB,eV)和价带电位(EVB,eV)可分别由下式计算出:

式中,χ为半导体的绝对电负性(4.72 eV),由第一电离能和电子亲和能的算术平均值算得;Ef为标准氢的自由电子能(约4.5 eV);Eg(eV)为半导体带隙.各样品的Eg,ECB和EVB电位的数值列于表2.
与纯g-C3N4相比,C掺杂g-C3N4的Eg变小.骨架C自掺杂g-C3N4的C3N4C1,C3N4C2和C3N4C3样品随着C掺杂量增大,形成的共轭π电子使价带升高,导带降低,Eg逐渐缩短,且均比无定形C自掺杂样品要小.而C3N4C4,C3N4C5和C3N4C6样品随着无定形C掺杂量的增多,Eg增大,这可能是无定形C过量聚合包裹g-C3N4所致.

Fig.3 UV⁃Vis DRS spectra(A),(ahν)1/2relative electron energy change diagram(B),N2adsorption⁃desorption isotherms(C),pore volume distributions(D)of the catalysts
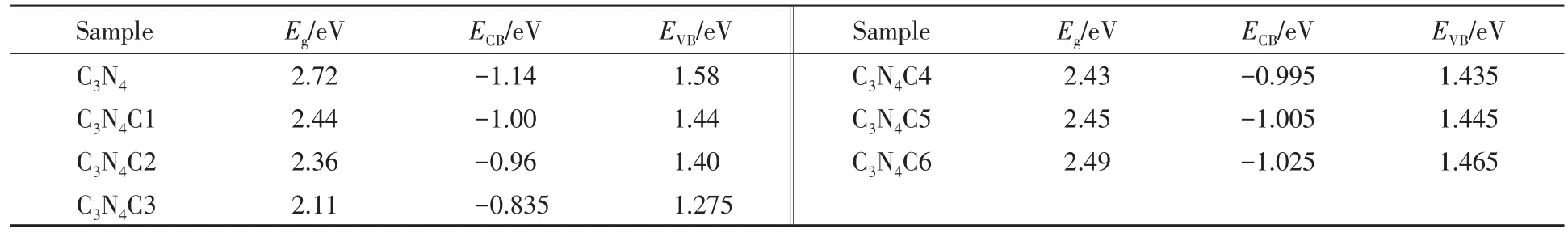
Table 2 Band gap,conduction band and valence band potentials of g-C3N4and C doped g-C3N4
样品的N2吸附-脱附等温线[图3(C)]均呈典型的Ⅳ型等温线,说明其为介孔结构.样品的孔径分布图[图3(D)]表明样品均具有多介孔结构,在3 nm处出现一个尖峰,在15~70 nm处出现一个宽峰,分别对应于g-C3N4纳米片的内腔直径和在g-C3N4纳米片之间形成的裂缝状孔.C3N4,C3N4C1,C3N4C2,C3N4C3,C3N4C4,C3N4C5和C3N4C6的BET比表面积分别为62.89,124.54,143.50,182.28,98.86,97.39和93.74 m2/g.由以上分析可知,随着C3N4C1,C3N4C2和C3N4C3骨架自掺杂C量的增多,样品的比表面积增大,3 nm中孔/介孔容积增大,6~70 nm的裂缝状孔大量存在.随着沉积到g-C3N4表层C量增大,无定形C会逐渐堵塞g-C3N4的中孔/介孔,故C3N4C4,C3N4C5和C3N4C6样品的比表面积和3 nm中孔/介孔容积逐渐减小.C自掺杂g-C3N4提升的比表面积有利于提供更多的活性位点吸收目标污染物,同时样品的多孔结构有助于反应物转移,这些有利因素都将大大提高光催化活性.
C自掺杂不仅可以调控g-C3N4的电子结构,而且对其微观形貌也有显著影响,图4示出了g-C3N4和C掺杂g-C3N4样品的形貌.可见,纯g-C3N4形貌为堆积不规则的光滑卷曲纳米片,具有丰富的石墨层状结构,卷曲片层成团聚堆积.随着C取代g-C3N4骨架中N原子的量增加,C3N4C1,C3N4C2和C3N4C3也呈分散薄层片状结构,片状卷曲度下降,薄层片状表面光滑且富有逐渐增强的刚性,片层上的孔洞增加.C3N4C4,C3N4C5和C3N4C6因C以无定形形式沉积负载在g-C3N4表面上,致使g-C3N4薄层片状逐渐减少,逐渐黏连聚合成块状结构.

Fig.4 SEM images for C3N4(A),C3N4C1(B),C3N4C2(C),C3N4C3(D),C3N4C4(E),C3N4C5(F)and C3N4C6(G)
2.2 骨架C掺杂g-C3N4和无定形C掺杂g-C3N4的形成机理
当单宁酸浓度≤10 mg/mL时,尿素具有大量的―NH基团,可作为氢键的给体和受体[21],单宁酸具有―OH基团,因此尿素分子之间、尿素分子和少量单宁酸分子之间可相互形成3D氢键超分子网络结构.在尿素聚合过程中,由于尿素具有氢键超分子结构,可以降低尿素的升华.在140℃左右,尿素汽化和分解成大量的NH3和氰酸,坩埚里面氧气会被消耗掉,形成富氮氛围[29].当温度超过220℃且持续升高时,氢键超分子结构中所捕获单宁酸分子会分解成CO2和碳化为取代g-C3N4骨架里面部分N原子形成骨架C掺杂.同时,单宁酸分解的CO2气体提供了一个气泡模板,防止g-C3N4纳米片堆积成团块,故能获得分散薄层片状结构的骨架C掺杂g-C3N4.当单宁酸浓度≥20 mg/mL时,尿素和单宁酸形成的3D氢键超分子网络结构中的单宁酸逐渐增多,基于单宁酸结构的多羟基性,会限制尿素分子间3D氢键形成和缩聚反应,以致尿素无法缩聚成片状g-C3N4,主要生成碎裂的g-C3N4,大量单宁酸碳化形成无定形C黏附沉积在g-C3N4碎片上面,聚合黏连成块状结构的无定形C掺杂g-C3N4.

Fig.5 Diagrams of(c/c0)⁃t(A)and ln(c/c0)⁃t(B)for photocatalytic degradation of RhB over catalysts under ultraviolet light irradiation
2.3 g-C3N4和C掺杂g-C3N4样品的光催化性能
图5和表3示出了紫外-可见吸收光谱法测定不同样品光催化降解RhB的催化性能数据.可知,骨架C自掺杂的C3N4C1,C3N4C2和C3N4C3随着C掺杂量的增大,光催化性能增强,降解率增大.无定形C掺杂的C3N4C4,C3N4C5和C3N4C6随着C掺杂量的增大,光催化性能减小,降解率减小.g-C3N4的骨架C自掺杂样品均比无定形C自掺杂样品的光催化性能要强.光照1 h后,C3N4C3的降解率达到89.07%,分别是纯g-C3N4和C3N4C4降解率的3.13倍和1.79倍.通过对降解率的数据进行拟合[图5(B)],光降解RhB符合一级反应动力学方程ln(c/c0)=kt,是一个拟一级反应过程[28].其中C3N4C3的光降解RhB反应速率常数分别是C3N4和C3N4C4的6.64和3.28倍.虽然紫外-可见光谱监测论证了光降解RhB符合拟一级反应过程,但这仅反映了RhB紫外光谱生色基团随时间的变化,而不能说明其后续中间产物的环裂解及氧化过程[30].
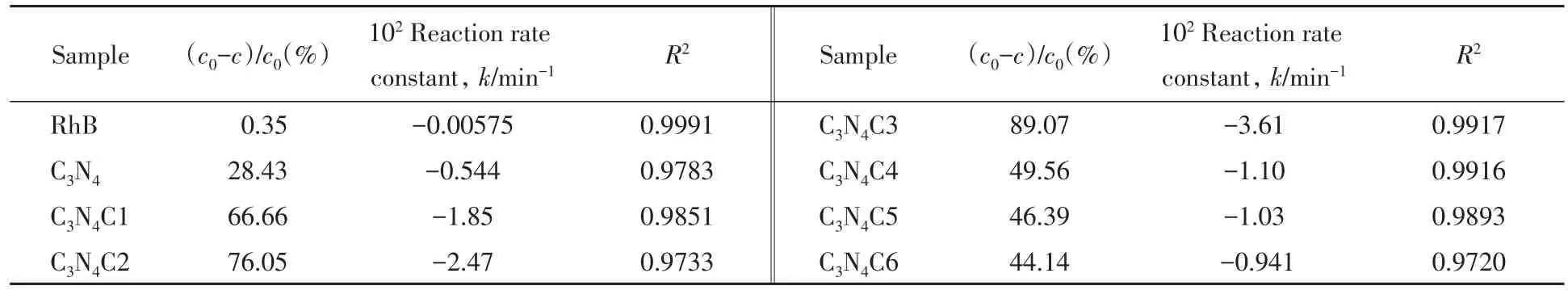
Table 3 Photocatalytic performance of rhodamine B degradation by light reaction of each sample for 1 h
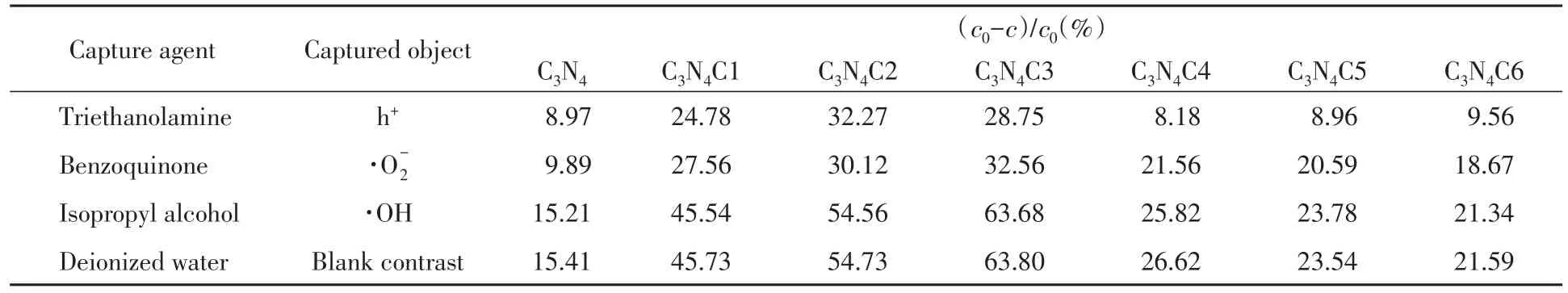
Table 4 Effects of different capture agents on photocatalytic degradation of RhB in C3N4,C3N4C1,C3N4C2,C3N4C3,C3N4C4,C3N4C5,C3N4C6

Scheme 1 Charge separation,transfer and degradation mechanism of RhB photocatalytic degradation by C3N4,C3N4C3 and C3N4C4
2.4 g-C3N4和C掺杂g-C3N4光降解RhB的微观机理
室温下不同俘获剂对各催化剂光催化降解RhB降解率的影响见表4,骨架C掺杂和无定形C掺杂的g-C3N4光催化降解RhB的机理见Scheme 1.由表4可知,三乙醇胺(h+)、对苯醌(·O-2)和异丙醇(·OH)[31]的加入对各样品光催化降解RhB反应均产生一定的抑制作用.其中加入异丙醇俘获剂对各样品光催化性能影响不大,与空白对照组基本一致,说明各样品光催化降解RhB的主要活性物种不是·OH.但当分别加入三乙醇胺和对苯醌后,C3N4,C3N4C1,C3N4C2和C3N4C3的光催化性能受到显著抑制,说明C3N4,C3N4C1,C3N4C2和C3N4C3光催化降解RhB的主要活性物种是h+和·.而C3N4C4,C3N4C5和C3N4C6光催化性能主要受到三乙醇胺抑制作用,因此其光降解RhB的主要活性物种是h+.这是因为各样品价带电子的电极电势均比E0(OH-/·OH)(pH=7,相对氢电极为2.31 eV)小,不能把OH—还原成·OH,故·OH不是各样品的主要活性物种.g-C3N4受光照后,电子从价带跃迁到导带,同时价带上产生相应空穴(h+),即产生光生电子和空穴.C3N4,C3N4C1,C3N4C2和C3N4C3导带光生电子的电极电势均比相对氢电极为-0.046 eV)值更负,所以能将O2还原为·,而聚集在g-C3N4价带上光生h+具有很强氧化性,能够直接氧化吸附在催化剂表面的RhB分子,故其主要活性物种是h+和·.C3N4C4,C3N4C5和C3N4C6样品是在g-C3N4表面原位沉积碳层,导带上的光生电子转移到了原位碳层上,增强了g-C3N4的电导性和电子转移能力[20],促进了电子-空穴分离,而聚集在g-C3N4表面上的空穴能够直接氧化吸附在催化剂表面的RhB分子,因此其催化性能比C3N4增强;但也因其原位碳层对导带电子进行转移,由导带光生电子氧化生成的·减少,故不及C3N4C1,C3N4C2和C3N4C3催化性能好.
结合UV-Vis DRS、PL、光催化性能结果、俘获剂实验以及g-C3N4能带结构分析,推测各样品光催化降解RhB机理如下.C3N4C1,C3N4C2和C3N4C3样品是骨架C掺杂g-C3N4,根据UV-Vis DRS结果分析,随着g-C3N4骨架C掺杂含量增加,样品禁带宽度缩短,有利于g-C3N4在可见光区域的光吸收,但价带位置上移,导带位置降低,分别使样品的氧化还原能力变弱.g-C3N4富含三均三嗪单元,其结构中C,N原子以sp2杂化形成高度离域的π共轭体系,其桥接N原子被C取代后会形成离域大π键,根据Li等[32]通过密度泛函理论研究骨架C掺杂g-C3N4的π电子浓度和电子结构发现,纯g-C3N4的HOMO轨道上的空穴和LUMO轨道上的电子均匀分布在三均三嗪环中,C掺杂后导致了HOMO轨道上的空穴和LUMO轨道上的电子进行空间分离,促进了g-C3N4上的电荷分离,降低了电子-空穴复合的几率,这是光催化性能提高的主要因素.C3N4C1,C3N4C2,C3N4C3样品随着C掺杂量增大,有更多的π电子促进光生载流子分离,这与PL结果一致.且根据BET和孔径分布图,C3N4C1,C3N4C2和C3N4C3的比表面积逐渐增大,孔隙率逐渐增大,有利于提供更多的活性位点吸收目标污染物和反应物转移.综合以上因素,C3N4C1,C3N4C2和C3N4C3样品随着C掺杂量增大,光催化性能增强.无定形C掺杂的C3N4C4,C3N4C5和C3N4C6样品随着C掺杂量增大,因其比表面积和孔隙率逐渐降低,且有大量C包裹住g-C3N4形成团聚,不利于光吸收,故催化性能逐渐降低.
2.5 g-C3N4和C掺杂g-C3N4光降解RhB的原位热动力学
图6(A)是各催化剂在298.15 K下光催化降解RhB的原位热谱曲线图.由图6(A)可知,各样品光催化降解罗丹明B的原位热力学曲线主要经历3个过程:从a到b为罗丹明B和催化剂分子的光响应吸热过程;从b到c为光响应吸热和光催化降解污染物放热的平衡过程;从c到d为光照反应1000 s后进入一个稳定放热阶段.在光催化反应1 h中,C3N4,C3N4C1,C3N4C2和C3N4C3样品的随着C掺杂量增大,ad阶段产生热效应逐渐增大,其中C3N4C3的热效应为3926.52 mJ,分别是相同条件下C3N4的2.65倍和C3N4C4的1.65倍.C3N4C4,C3N4C5和C3N4C6样品随着C掺杂量增大,ad阶段产生热效应逐渐减小.在相同的光催化时间内,如果降解过程放出的热量越大,说明单位时间的放热速率越快,光催化效果越佳.
光催化降解速率Rt可根据下式由反应热变速率(dQt/dt)求出:

式中,dQt为t时间反应热(mJ);ΔQtheor0为光催化完全降解罗丹明B时理论反应热(mJ).
光催化降解罗丹明B的热变曲线(Et-t)与热功率的关系见下式:

式中,Pt是t时间的热功率(mW);Et是原位量热曲线中t时刻的热电势(μV);S为298.15 K下光量热计的热量参数,为(66.85±0.56)μV/mW.
结合焦耳定律Q=W=Pt×t,热变速率可由下式求出,由此获得C3N4,C3N4C1,C3N4C2,C3N4C3,C3N4C4,C3N4C5和C3N4C6在298.15 K下光催化降解罗丹明B的热变速率曲线[图6(B)]和原位热动力学数据(表5).

由图6(B)可知,各样品的热变速率曲线与电势曲线变化特征基本一致,整个反应最终进入一个稳定放热平台阶段,各样品在cd阶段放热速率数值详见表5,C3N4C3在稳定的cd放热阶段的热变速率分别是C3N4的1.84倍和C3N4C4的1.5倍.在这个阶段单位时间内被激活的催化剂数量恒定,单位时间内产生的·和光生h+的浓度基本趋于恒定,因此光催化降解速率趋于恒定,这决定了此阶段是一个拟零级决速过程.Gaisford等[33]研究表明光量热法是一种用于研究硝苯吡啶光分解零级过程的通用方法,在一定功率的光照条件下,单位时间内反应的硝苯吡啶分子数量是相同的,与溶液的体积和初始浓度无关.
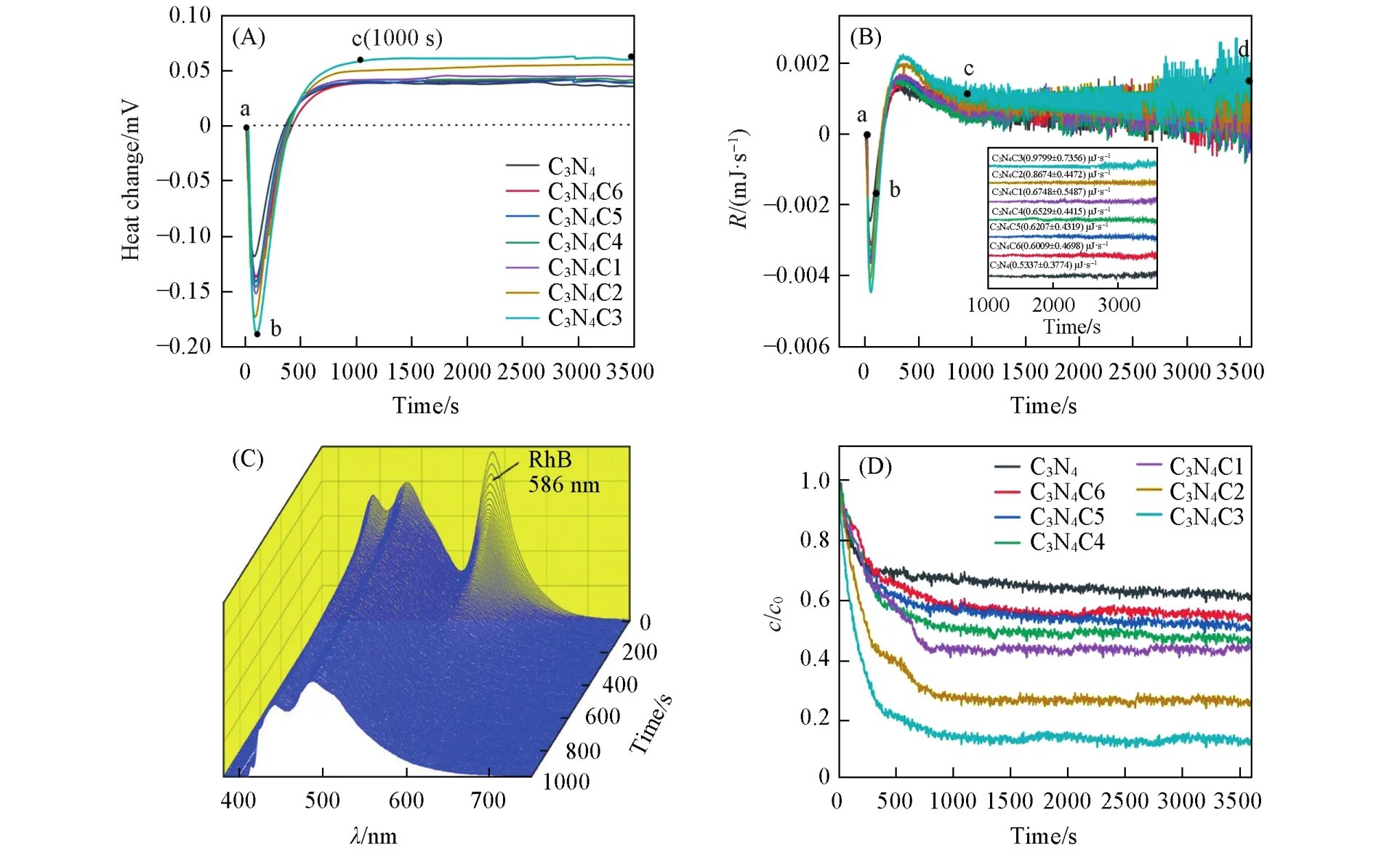
Fig.6 In situ heat change curves(A)and in situ heat flow rate curves(B)of photocatalytic degradation of RhB by C3N4,C3N4C1,C3N4C2,C3N4C3,C3N4C4,C3N4C5,C3N4C6,in situ 3D fluorescence spectrogram(C)of photocatalytic degradation of RhB by C3N4C3 under 298.15 K and the c/c0curve of the degra⁃dation process of RhB with time measured by in situ 3D fluorescence spectrogram(D)

Table 5 In situ thermodynamic/kinetic data of Rhodamine B photocatalytic degradation
图6(C)是用光微量热-荧光光谱联用仪测试C3N4C3在298.15 K光催化降解罗丹明B的原位三维荧光光谱图,586 nm是由罗丹明B的发色基团产生的荧光峰,而442和482 nm是由g-C3N4产生的峰.随着光照时间的延长,罗丹明B分子在586 nm处的特征荧光发射峰强度开始剧减,光照1000 s后,586 nm处荧光发射峰的强度不再变化.图6(D)是采用光微量热-荧光光谱联用仪测试各样品在光催化过程中RhB降解率随时间的变化谱图.光反应开始后,各样品RhB降解率骤减,对应图6(A)热流曲线的ac阶段,这是因为光反应开始后,光照产生的活性物种作用于RhB分子,破坏了RhB及含荧光发色基团的中间产物,导致586 nm处荧光发色基团的荧光强度骤减.光反应1000 s时,各样品的RhB降解率达到一个稳定值,其中C3N4C3的RhB降解率为87.6%,分别是C3N4和C3N4C4的2.65倍和1.95倍.且对应各热流曲线中的稳定放热平台cd阶段,说明各样品光催化反应并未终止,主要是以降解非荧光发色基团的中间产物,包括中间产物矿化及含苯环的中间产物开环等为主,此阶段降解速率很慢,是光催化反应决速步骤.因此,利用光微量热-荧光光谱联用仪研究碳掺杂g-C3N4光催化降解RhB过程是一个拟零级反应过程而非紫外-可见光谱显示的拟一级反应动力学过程.
3 结 论
通过在尿素前驱体中调节单宁酸的用量,原位缩聚形成骨架C掺杂g-C3N4和无定形C掺杂g-C3N4,对C掺杂g-C3N4的物相形貌结构和能带价态组分进行表征分析,结合紫外-可见吸收光谱和原位光微量热-荧光光谱联用仪获取了碳自掺杂g-C3N4降解罗丹明B的原位热/动力学信息和三维荧光光谱信息,探讨了2种掺杂方式的光催化性能和光催化降解罗丹明B的微观机制.结果表明,单宁酸浓度≤10 mg/mL时,碳会取代七嗪单元结构的N原子形成g-C3N4骨架碳自掺杂;单宁酸浓度≥20 mg/mL时,碳以无定形形式沉积负载在g-C3N4表面上形成无定形碳自掺杂.相比原始g-C3N4和无定形碳自掺杂g-C3N4,骨架碳自掺杂g-C3N4形成的π电子有效缩短了禁带宽度,减小了光生电子-空穴复合几率,显示出优异的光催化性能,光催化主要活性物种为h+和·.紫外-可见吸收光谱检测结果显示,骨架碳掺杂g-C3N4(C/N=0.844)的RhB降解率达到89.07%,分别是原始g-C3N4和无定形碳自掺杂g-C3N4的3.13倍和2.02倍.光催化降解过程可分为罗丹明B和催化剂光响应吸热过程、光响应吸热和光催化降解污染物放热的平衡过程与稳定放热这3个过程.其中在骨架碳掺杂g-C3N4(C/N=0.844)下,光照1000 s后,三维荧光光谱检测的RhB降解率为87.6%,分别是原始g-C3N4和无定形碳自掺杂g-C3N4的3.13倍和1.95倍.光照1000 s后,光微量热计显示以矿化和降解非荧光发色中间产物为主,并保持以热变速率为(0.9799±0.5356)μJ/s稳定放热,为拟零级反应过程,是光催化反应决速步骤.碳掺杂g-C3N4光催化降解RhB过程是一个拟零级反应过程而非拟一级反应动力学过程.
