荧光蛋白结构改造及其生物传感应用
2020-11-13李慧圆雷春阳
李慧圆,雷春阳,黄 燕,聂 舟
(湖南大学化学化工学院,化学/生物传感与化学计量学国家重点实验室,湖南省生物大分子化学生物学重点实验室,长沙410082)
新的技术和研究工具的出现,通常可以加速甚至彻底改变生物学研究.在过去几十年中,荧光蛋白的出现和最先进的显微技术使人们进入一个可以在细胞或亚细胞分辨率下观察活细胞的细微结构和原位实时监测多种生理、病理过程的全新阶段[1].其中,来自维多利亚多管水母(Aequorea victoria)的野生型绿色荧光蛋白(Wild-type green fluorescent protein,wtGFP)是第一个被克隆并进行表达的荧光蛋白[2].wtGFP因其独特的性质和稳定的结构吸引了大量科研工作者对其开展相应的研究.鉴于荧光蛋白在现代生命科学研究中的重要性,在荧光蛋白的发现和应用过程中做出杰出贡献的3位科学家Osamu Shimomura,Martin Chalfie和Roger Y.Tsien被授予了2008年诺贝尔化学奖[3].
1955年,Davenport和Nicol[4]首次发现Aequorea victoria在紫外光照射下可以发出绿色的荧光.1962年,Shimomura等[5]在这类水母中首次发现绿色荧光蛋白(GFP).他们进一步通过木瓜蛋白酶将GFP水解成多肽片段,并分离、纯化含有生色团的多肽,推测出GFP生色团的化学结构为4-对羟基苯甲基-5-咪唑啉酮[4-(p-hydroxybenzylidene)-5-imidazolinone,p-HBI][6]. 此时,研究者并未意识到GFP的巨大应用潜力.1992年,Prasher等[7]克隆并测序确定了wtGFP的cDNA序列,科学家们开始意识到它的重大意义.1994年,Chalfie等[8]实现了GFP在异源生物体线虫中的表达,开启了GFP作为荧光遗传标签的时代.Tsien[9]是第一位致力于荧光蛋白改造的科学家,他和团队通过定点突变和随机突变获得了新的GFP衍生物,并阐述了GFP的发光机制,为同时追踪多个生理事件提供了有力的技术支持.1999年,Matz等[10]从珊瑚虫中发现并分离出编码红色荧光蛋白(DsRed)的基因.虽然DsRed是一种不利于与其它蛋白进行融合表达的四聚体蛋白,但这一发现仍是鼓舞人心的.2002年,Campbell等[11]通过定向进化的方式在DsRed的基础上得到了单体红色荧光蛋白(mRFP1).在众多科学家的努力下,目前已报道的荧光蛋白的荧光光谱涵盖了整个可见光区乃至近红外区域,为活细胞内可视化和量化各种蛋白和生理事件提供了丰富的工具[1].
对荧光蛋白分子结构的解析为揭示其发光机制及更广泛地应用于生命科学研究领域奠定了基础.wtGFP包含238个氨基酸,分子量约27000.晶体结构分析结果表明,该蛋白质是由11个β-折叠围绕着沿中心轴延伸的α-螺旋组成的桶状结构(β-桶),α-螺旋上的三肽(Ser65-Tyr66-Gly67)通过自催化分子内环化反应生成生色团.生色团被封装在桶状结构的中心,短的α-螺旋段盖住桶的两端,有助于将生色团与溶剂隔离开.生色团的成熟包括折叠-环化-脱水-氧化4个过程,除O2外不需要其它辅因子.最初的折叠步骤至关重要,只有当GFP多肽链正确折叠成稳定的天然构型时,才能进行下一步的生色团形成[12].
通过对荧光蛋白结构和成熟过程的深入探究,越来越多的研究者着手对荧光蛋白结构进行重新设计,赋予其新的功能和性质,这进一步促进了荧光蛋白的发展与应用.本文将系统介绍通过局部结构改造、桶状结构重构、表面重构和结构模拟等结构设计方法实现对GFP功能的改变和模拟,并且介绍不同结构改造方法获得的荧光蛋白在生物传感方面的代表性应用,为探究其它荧光蛋白的改造及在生命科学领域的应用提供了设计思路.
1 局部结构改造
wtGFP具有复杂的光谱性质(λex:397 nm/λem:508 nm,λex:476 nm/λem:503 nm)以及低的量子产率,严重限制了其在生物成像方面的应用[13].科学家们希望可以获得更明亮、更稳定以及不同颜色的荧光蛋白(图1).最初,较广泛的尝试是通过定点突变技术对wtGFP进行结构改造,来探究是否可以使用不同的氨基酸对特定位点进行替换以调整其光谱特征.
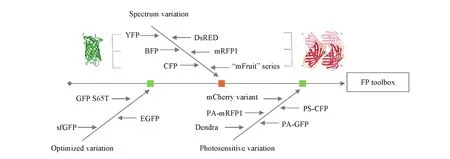
Fig.1 Schematic representation of the fluorescent protein mutants
Tsien[9]通过定点突变的方法首先对临近wtGFP生色团位置的氨基酸进行突变,获得了一系列发射光谱位于蓝色到黄色区域的荧光蛋白突变体,包括蓝色荧光蛋白(BFP和GFP Y66H,λex:380 nm/λem:448 nm)[14]、青色荧光蛋白(CFP和GFP Y66W,λex:380 nm/λem:448 nm)以及黄色荧光蛋白(YFP,GFP S65T和T203H,λex:512 nm/λem:524 nm)[15]. 不同颜色的荧光蛋白突变体为多色成像以及构建基于荧光蛋白的荧光共振能量转移(FRET)传感器提供了可能.Rizutto等[16]首次使用BFP和GFP S65T对亚细胞结构实现了双重标记.Tsien课题组[17]完成了第一个基于荧光蛋白的FRET实验,而后成功获得了检测Ca2+的FRET传感器cameleons[18].这一突破性的设计可以监测单个细胞中游离Ca2+的信号,表明基于荧光蛋白的FRET生物传感器在细胞成像中具有广泛的应用潜力.CFP/YFP组合是迄今为止绝大多数基于荧光蛋白FRET生物传感器的报告元件,被用于蛋白激酶活性、caspase-3活性、钙离子、锌离子及稀土元素等的检测[19~23].除了通过突变获得多色荧光蛋白外,研究者也在寻找与增强型绿色荧光蛋白(EGFP)具有相似性能的红色荧光蛋白.Campbell等[11]通过定向进化替换了DsRed中的33个氨基酸,获得mRFP1.此后,科学家们进一步获得了“mFruit”系列的荧光蛋白,其中包括mOrange,tdTomato,mStrawberry,mCherry和mPlum等荧光蛋白[24].这些多色荧光蛋白突变体的出现很好地补充了wtGFP突变体成像区域的空白.
此外,通过定点突变技术对特定位点氨基酸进行替换也可有效改善荧光蛋白的折叠效率,提高GFP在不同的异源细胞系统中的表达效率[25]. 如,GFP S65T(λex:489 nm/λem:510 nm)稳定了发色团的氢键网络,亮度提高约5倍,成熟速度提高约4倍,改善了GFP作为报告蛋白存在的限制[13].EGFP(GFP S65T和F64L)在37℃时的成熟效率极高[26],在生物成像领域发挥着重要的作用[27].Waldo课题组[28]对wtGFP的16个氨基酸残基进行突变,获得了超折叠绿色荧光蛋白(sfGFP).sfGFP具有更加稳定的荧光性质,即使与不溶性蛋白融合也能折叠,为后续荧光蛋白的结构设计和改造奠定了基础.
许多荧光蛋白的光物理特性非常复杂,可能涉及几种不同的发射和非发射状态,以及在单个分子水平上观察到的开关“闪烁”行为[29].在wtGFP及其增强型突变体中,研究者首次观察到了光敏现象,为研究活细胞成像中蛋白质动力学过程提供了有利工具[30],使对目标蛋白进行精确的光标记和跟踪成为可能.如,光激活变体PA-GFP(GFP H203T)在413 nm光预照射下会发生光转换,从而在488 nm光激发下具有更明显的光学对比度(增加约100倍的绿色荧光),可以实现对标记蛋白移动的直接观察且有效避免了新合成荧光蛋白的信号干扰[31].源自墨绿多管水母(Aequorea coerulescens)的aceGFP的光转换突变体PS-CFP在405 nm光照射下会从青色(468 nm)转变为绿色荧光(511 nm),很好地解决了光激活突变体在激活前无法确定激活区域的问题,成功用于活细胞中多巴胺转运体的转运研究[32].随后,一些研究小组通过定点突变技术也发展了一系列基于红色荧光蛋白的光敏蛋白突变体,包括光激活红色荧光蛋白PA-mRFP1、mCherry可光活化变体及Dendra等,为活细胞的多色光激活标记和超分辨率显微镜研究储备了工具[33~35].
目前,对常用的荧光蛋白大多通过定点突变和定向进化技术进行局部结构改造,以改善荧光蛋白的光谱特性、光稳定性、成熟效率和亮度,提高了这类蛋白用于细胞成像的适用性.但这种方式通常具有较大的随机性,需要构建较多的突变体并且需要经过大量筛选以获得最终具有良好性质的荧光蛋白.
2 结构重构
蛋白质需要完整的结构和正确的折叠才能获得相应的生物学功能.许多蛋白被劈裂成2个片段后,通过复合还可以重新形成具有生物学功能的复合物,包括泛素、β-半乳糖苷酶和二氢叶酸还原酶等[36~38].Richards[39]最早发现牛胰核糖核酸酶被枯草杆菌蛋白酶切割后形成的多肽和残留蛋白具有较低的活性(不到酶原始活性的5%),当多肽和残留蛋白混合后,能重新复合为具有功能的复合体,据此首次构建了基于酶的蛋白互补对.
随着对荧光蛋白的结构-功能的深入研究,除了通过突变的形式对荧光蛋白进行结构改造之外,研究者还通过结构重构的方式对荧光蛋白的结构进行了重排和重组.如通过循环重排对其C,N端位置进行重新设置、在特定位点插入外来片段或进行劈裂后再进行复合,荧光蛋白均可以正确地折叠并发出荧光.1999年,Baird课题组[40]通过循环重排研究了GFP的突变体(ECFP,EYFP和EGFP),对EGFP的所有位点进行筛选,发现10个可以进行切割的位点,分别位于第7,8和11个β-链,或者位于7和8,8和9β-链之间的loop区域.这些位点的循环重排或外源多肽的插入不会影响GFP的光谱性质,也成为后来结构重构中分割荧光蛋白的位点.其中,荧光蛋白结构重构中最初代表性的技术就是双分子荧光互补技术(BiFC).Regan课题组[41]最早实现了基于GFP的蛋白互补.他们将1个GFP突变体(sg100)在蛋白的第7和第8个β-链loop区域进行分割,产生2个大小相近的片段(N-GFP和C-GFP);分割后的GFP片段只有通过具有强烈相互作用的反平行亮氨酸拉链的作用才能重新组装成完整的GFP并发出荧光.之后,Kerppola课题组[42]提出了BiFC的概念,对GFP和其它颜色的GFP突变体的BiFC现象进行了系统研究,并通过多色BiFC同时鉴定了多种活细胞中蛋白质的相互作用[43].基于BiFC的检测具有简单直观的特点,已经被广泛应用于生物大分子间相互作用的可视化检测[44,45].然而,最初得到的BiFC体系对温度很敏感.此外,蛋白大片段与目标蛋白融合表达时,可能会影响融合蛋白的正确折叠[46,47].
在sfGFP的基础上,Waldo等[48]在10和11β-链loop区域确定了一个合适的切割位点,发展了“10+1”分裂体系.相对于“7+4”和“8+3”体系,“10+1”体系不需要借助外界的相互作用即可自发复合且不影响融合蛋白的折叠.他们进一步用该体系实现了对蛋白可溶性表达的评估.在此基础上,Brock等[49]基于“10+1”体系开发了一种可视化分析穿膜肽内吞过程的分析方法.Ozawa课题组[50]开发了一种可视化检测活体内细胞凋亡的蛋白探针,通过基因工程将GFP11小片段与细胞凋亡有关的Smac蛋白进行融合[图2(A)].当细胞受到凋亡信号刺激时,带有GFP11小片段的Smac蛋白被释放到细胞质中,启动细胞凋亡机制,同时GFP1-10蛋白通过载体质粒在细胞质中表达,将会与释放出来的Smac蛋白上的GFP11发生复合重构而产生荧光信号,从而实现可视化监测细胞凋亡过程.
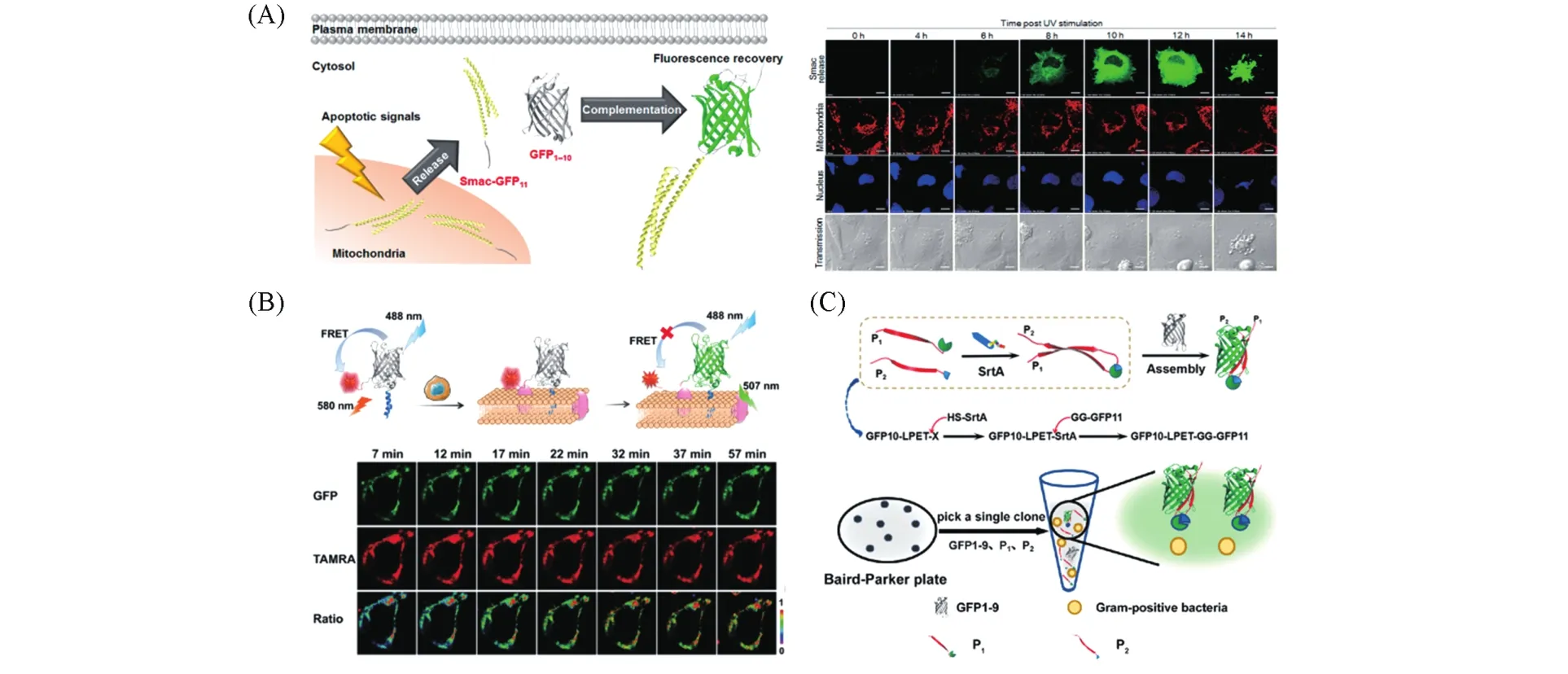
Fig.2 Representative applications of split fluorescent protein in biosensing
“10+1”分裂体系有效地补充了荧光蛋白结构重构策略,并且小片段标记对目标蛋白的折叠效率和溶解度影响不大.将GFP11与目标蛋白的N或C末端融合表达,通过GFP1-10和GFP11的自组装,可以实现目的蛋白的标记[48].此外,小片段还可以通过多肽固相合成的方法获得,序列可设计性强,且容易引入化学修饰,大大拓展了“10+1”分裂体系的应用范围.基于此,我们[51]设计了基于半合成荧光蛋白功能化的FRET探针(FPAP)[图2(B)].连有小分子FRET受体和蛋白酶底物序列的GFP11可以和GFP1-10片段自发复合,形成的探针通过GFP1-10片段上修饰的膜插入肽(MIP)锚定在活细胞膜上,细胞膜上的目标酶通过切割底物序列释放出小分子FRET受体,从而实现对活细胞膜上弗林蛋白酶催化反应动态过程的原位实时成像.此外,我们[52]还提出了一种基于羧肽酶的磷酸化保护半合成GFP自组装的策略,实现了对蛋白激酶活性的检测.化学合成的s10包括GFP10片段和磷酸化底物多肽,蛋白激酶催化的底物磷酸化可以保护GFP10片段不被羧肽酶切割,使其可以与带有成熟生色团的截短蛋白(tGFP)自组装并恢复生色团荧光,呈现比传统分裂GFP系统更快的荧光互补和更高的信倍比.
Waldo课题组[53]进一步对双分裂体系进行改造,设计了三分裂GFP体系.该体系包括GFP1-9,GFP10和GFP11三个部分(9+1+1),三者不能自发复合形成完整的GFP.只有当GFP10和GFP11片段因伴侣蛋白之间的相互作用而靠拢时,才会与GFP1-9大片段发生复合形成具有完整结构的GFP,并伴随荧光信号的产生.三分裂GFP体系同样能够检测蛋白-蛋白相互作用[54].Cabantous等[55]基于三分裂GFP体系开发了小G蛋白(GTPase)及其效应因子相互作用的传感器,实现了对GTPase激活调节因子的高通量筛选.同样的,三分裂体系中的小片段也具有不干扰目的蛋白折叠、易于设计的特点.我们[56]建立了一种基于三分裂GFP体系的转肽酶活性检测新方法[图2(C)].化学合成的GFP10和GFP11片段包含转肽酶识别序列,在转肽酶的催化下通过转肽反应形成GFP10-11片段,从而与GFP1-9复合形成完整的GFP并伴随产生荧光信号.该方法无需标记且灵敏,其简单的“一锅法”操作过程可用于实际样品中金黄色葡萄球菌的检测.近期,Shu等[57]设计了一种名为FlipGFP的蛋白酶活性传感器,按GFP10,E5,GFP11,蛋白酶的切割序列和K5的顺序依次将这些多肽进行融合并表达,异二聚线圈E5和K5的作用反转了GFP11相对于GFP10的方向,产生具有同向平行结构的GFP10-GFP11链.当蛋白酶切割序列后可还原GFP11与GFP10的反平行结构,并与GFP1-9结合并恢复荧光,从而实现蛋白酶活性的灵敏检测.作者将TEV切割序列插入到FlipGFP中,在TEV切割序列后获得77倍的荧光变化;通过将切割序列更换为caspase-3的切割序列,实现了对细胞凋亡过程的监测.
对荧光蛋白进行结构重构的方法为研究生物大分子相互作用提供了新思路.但劈裂位点的选择、复合之后荧光蛋白荧光强度以及成熟速率的提高还需要进一步研究.
3 表面重构
蛋白表面重构是通过计算机模拟和遗传改造的方法对蛋白质表面进行重新设计,可在不破坏蛋白质原有功能和性质的基础上,扩展蛋白质的功能和性质[58].Liu等[59]通过表面重构策略对GFP进行改造,探究了蛋白抗聚集性与表面净电荷之间的关系.他们将sfGFP中的表面氨基酸残基突变成精氨酸/赖氨酸(Arg/Lys)或谷氨酸/天冬氨酸(Glu/Asp),获得了理论上带大量净电荷(正/负)的超电荷荧光蛋白(ScGFP),并成功在大肠杆菌中表达出这些蛋白.实验结果表明,ScGFP拥有和起始GFP相似的光物理性质,并且具有抗聚集和强大的热稳定性等优异性质.
ScGFP对带相反电荷的大分子表现出强大、可逆的亲和力,这类似于组蛋白与带相反电荷的DNA的天然组装[60].受DNA/组蛋白复合物自然装配行为的启发,我们[61]将+36 GFP和带负电的DNA通过静电相互作用组装形成聚离子复合物,开发了一种基于+36 GFP/DNA聚离子复合物和支点介导链取代反应的生物传感新方法[图3(A)].该方法中,双链DNA探针包含长链(L)和短链(S),S链尾部用有机猝灭剂BHQ1进行修饰.当BHQ1-DNA与+36 GFP形成纳米复合物时,前者通过能量转移高效猝灭+36 GFP的荧光.当与L链完全互补的靶DNA(T1)存在时,会与探针发生支点介导的链置换,从而释放BHQ1标记的S链,并恢复+36 GFP荧光;当目标链中含有突变位点时,链取代不会发生,荧光不能恢复,从而构建了一个用于基因诊断的简便通用的平台.与分子信标探针相比,基于ScGFP的DNA分析显示出高灵敏度和更好的碱基错配区分能力,可应用于癌症病人组织样品中目标基因甲基化程度的快速检测.ScGFP除了可以与带负电的核酸发生相互作用之外,还可以与其它材料发生相互作用.通过研究DNA保护的+36 GFP与石墨烯(GO)的相互作用,我们[62]开发了DNA损伤相关酶活性检测的新方法,实现了尿嘧啶-DNA糖基化酶(UDGs)碱基切除修复酶的活性和抑制剂的分析检测.我们[63]还发展了基于+36 GFP与阳离子聚合物共振能量转移的荧光策略,实现了对多聚腺苷酸二磷酸核糖聚合酶(PARP-1)活性和抑制剂的高灵敏检测分析.同样,Li等[64]基于糖胺聚糖(GAG)剂量依赖性地抑制GO对+36 GFP的荧光猝灭效应,实现了从分子水平到细胞水平甚至活体的复杂生物样品中GAG的检测和成像.相对于需要遗传构建的传统生物传感器,基于ScGFP构建的生物传感方法具有方便快捷、灵敏以及响应机制多样化的特点.
许多阳离子型递送载体通常会与细胞膜上带负电荷的成分结合,并通过胞吞的方式进入细胞,如HIV Tat细胞穿透肽和有阳离子区域的工程蛋白等[65,66].研究者推测+36 GFP也可能与细胞膜的负电荷成分缔合从而穿透细胞.Liu等[67,68]验证了+36 GFP穿透细胞的能力,并进一步用+36 GFP将核酸和蛋白质递送到各种哺乳动物细胞内,包括对阳离子脂质体转染具有抗性的细胞系.+36 GFP很好地解决了大多数递送方法存在的普遍性问题,如细胞毒性和稳定性等,提供了一种高效无毒快速的生物大分子递送方法.
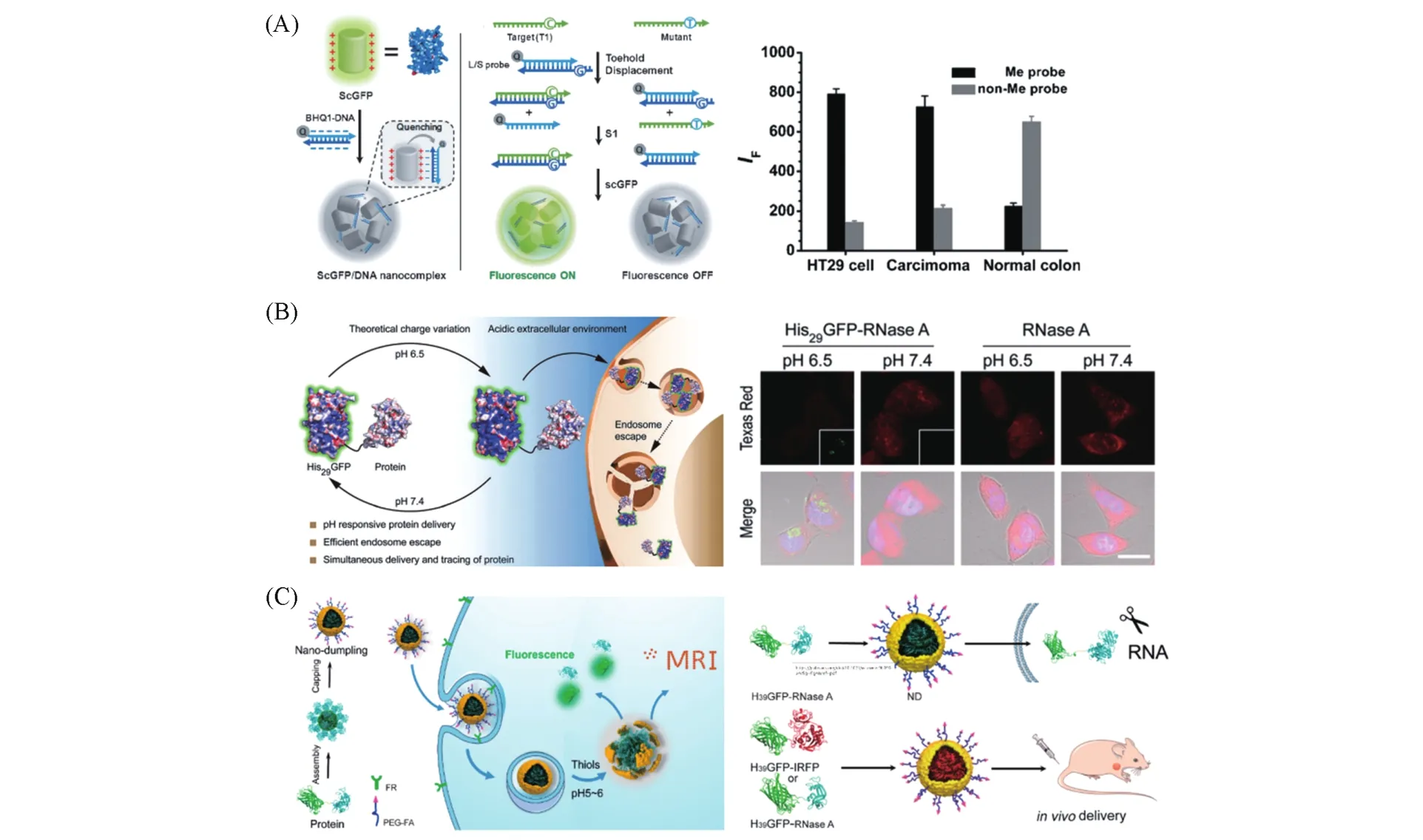
Fig.3 Representative applications of the surface reconstructed fluorescent proteins in biosensing and protein delivery
尽管+36 GFP的细胞转运效率高,但通过胞吞进入细胞的大多数蛋白仍被包裹在囊泡内,不是被溶酶体降解,就是再次被排出细胞,真正释放发挥作用的蛋白比较少.Liu等[60]通过表面重构的方法进一步对+36 GFP进行改造,将Arg/Lys突变位点替换成组氨酸(His),获得了表面带39个His残基的GFP(H39GFP),并且发现pH可以调节H39GFP表面所带的电荷量.我们[69]基于H39GFP表面电荷可调的性质构建了H+和金属离子触发的细胞逻辑门,用于核酸分子的智能递送,实现了条件可控的siRNA胞内递送及细胞内目标蛋白表达水平的下调.在此基础上,我们[70]设想通过调节His与Arg/Lys的比例,将富含His的GFP设计成准确响应目标pH的蛋白载体.根据肿瘤细胞微环境的pH值,通过表面重构进一步设计获得了His29GFP,该蛋白可以区分肿瘤和正常细胞微环境的微弱pH差异[图3(B)].His29GFP在目标pH=6.5时可选择性渗透细胞,并高效地从溶酶体中逃逸,从而将核糖核酸酶A(RNase A)递送至肿瘤细胞内,降解mRNA并引起细胞死亡.近期,我们[71]还将H39GFP与纳米材料结合,构建了蛋白药物负载量高(质量分数>63%)以及可实时监控蛋白药物释放的荧光/核磁双模成像的“纳米饺子”[图3(C)].融合了蛋白药物的His39GFP通过金属离子和组氨酸的作用引导蛋白自组装形成复合物作为“饺子馅”,在其表面通过二氧化锰(MnO2)的原位生物矿化形成“饺子皮”,进一步通过叶酸修饰的聚乙二醇(PEG-FA)进行功能化,获得主动靶向癌细胞的蛋白递送“纳米饺子”.MnO2纳米材料广泛的光吸收能力使其能够通过能量转移猝灭His39GFP的荧光,内涵体/溶酶体内还原性和酸性微环境的双刺激可以破坏MnO2纳米层,释放大量带有His39GFP标签的蛋白药物和Mn2+.结合蛋白载体和纳米材料载体的特性,该工作提出了一种高效智能的自报告蛋白递送新方法,实现了蛋白药物的高效靶向递送,并通过荧光/磁共振成像(MRI)对蛋白药物的释放过程进行实时跟踪,对于提高抗肿瘤蛋白药物的靶向性和药效具有重要的理论和应用价值.此外,利用H39GFP具有的金属离子配位性质,我们[72]设计了一种免标记检测Cu2+的传感方法,实现了对Cu2+的快速、简便和特异性检测.在此基础上,开发了基于His39GFP/Cu2+检测乙酰胆碱酯酶(AChE)活性的传感策略.
通过表面重构对荧光蛋白进行改造,可以赋予荧光蛋白独特的性质,丰富其在生物传感等生命科学领域的应用.表面重构的设计策略需要通过合适的方法筛选出可以突变的表面位点.与掩埋残基相比,溶剂暴露的氨基酸通常被认为对稳定折叠状态的贡献较小,但需要注意的是,无论溶剂暴露程度如何,侧链都可能在某些蛋白质的表达和折叠中发挥重要作用.
4 结构模拟
化学合成的荧光蛋白生色团HBI几乎没有荧光,这与在变性的GFP中观察到的荧光响应的快速消失一致.然而,一旦GFP重新折叠,荧光就会恢复.因此,生色团周围的环境对于荧光蛋白保持荧光至关重要[73,74].在GFP生色团附近存在着大量含有极性基团侧链的氨基酸残基,这些侧链与生色团形成氢键网络,从而限制生色团的分子内运动,使荧光成为消耗激发态生色团能量的主要途径[75].
受GFP结构和生色团发光机理的启发,研究者利用超分子宿主、金属有机框架、蛋白质和核酸模拟了GFPβ-桶的自然限制作用来限制生色团的分子内运动,从而实现了GFP生色团类似物的荧光激活[76~80].同时,利用GFP生色团类似物,发展了一系列“turn on”型的传感器,实现了对离子、核酸和蛋白质等目标物的检测[81,82].
相对于其它荧光蛋白结构模拟物,核酸模拟物除具有易设计合成的特点外,还为荧光标记核酸提供了合适的方法.基于荧光蛋白核酸模拟物的传感器不仅有效地补充了荧光蛋白传感器在核酸应用领域的不足,也促进了核酸在生物学研究领域的应用.三苯基甲烷染料,如孔雀石绿(MG),容易发生振动激发而通常具有极低的荧光量子产率.2003年,Tsien等[83]证明了MG与核酸适配体结合时会显著增加MG的荧光(约2360倍),这一开创性工作催生了可与各种弱荧光分子结合以增强这些分子荧光的RNA适体的发展[84~86].Jaffrey课题组[80]通过筛选对GFP生色团类似物具有足够亲和力和选择性的核酸适配体,开发了遗传编码的“荧光RNA”,得到了一系列光谱跨越可见光区的RNA-生色团复合物,其中一种叫作菠菜(Spinach)的RNA-生色团复合物发出的绿色荧光与GFP的亮度相当.随后,该课题组[87]将Spinach和目标识别适配体结合,利用目标物的结合诱导调节Spinach的结构,使之能与生色团类似物3,5-二氟-4-羟基苯甲基咪唑啉酮(DFHBI)结合并产生荧光,构建了在体外检测各种小分子的变构Spinach传感器,实现了对大肠杆菌中二磷酸腺苷(ADP)和S腺苷甲硫氨酸(SAM)的细胞内水平的动态变化和细胞间变化进行成像[图4(A)].基于此原理,通过对RNA序列结构进行重新设计,研究者开发了分别针对代谢物和蛋白质的各种Spinach传感器[88~91].
此外,Spinach传感器也可以对体外转录进行实时监测[92].Ricci等[93]通过对Spinach适体进行劈裂,开发了一种生物分子互补测定法.2条带有抗原标签的RNA互补链与靶抗体结合后共定位在狭窄的空间中,可以重新组装成Spinach构象,产生抗体浓度依赖的荧光发射.该方法为Spinach传感器的设计提供了新思路.核糖开关是天然存在的代谢物敏感RNA,对于构建传感器是一个不错的选择.Jaffrey课题组[94,95]利用细菌核糖开关中的适体,结合变构Spinach传感器的概念,开发了另一类基于荧光蛋白RNA模拟物的Spinach核糖开关,并应用于代谢物的检测.此外,该小组还发展了其它荧光蛋白RNA模拟物,包括Spinach2、花椰菜(Broccoli)和玉米(Corn)[96~98].Spinach2在荧光标记活细胞中的RNA方面展现出更好的热稳定性,Broccoli和Corn结合其相应的生色团类似物后分别显示绿色和黄色荧光.其中,Broccoli在细胞成像中显示出更加稳定的折叠和绿色荧光,并且相对于Spinach具有更小的尺寸和更强的荧光.这些荧光蛋白RNA模拟物的出现使得对活细胞中重要的信使、核糖体和微小RNA的原位荧光标记和成像成为可能,从而有助于回答有关其在活细胞中的代谢、运输和功能的许多生物学问题.
基于荧光蛋白RNA模拟物的生物传感器能在细胞中表达,并表现出与特定细胞内代谢物浓度成比例的荧光,已被用作代谢产物成像的遗传编码RNA传感器以及合成生物学应用的工具[99].然而,因为标记RNA的快速降解和热不稳定性等原因,这类RNA模拟物在哺乳动物细胞中的亮度有限.G-四链体(G4)是基于Hoogsteen碱基配对的4个G形成的核酸结构.研究表明,Spinach具有丰富的鸟苷酸(G)序列,包含一个独特的G4结构以促进生色团荧光的激活[100].基于此,我们[101]合成了一系列红色荧光蛋白(RFP)生色团类似物,并利用DNA G4点亮了这些生色团,首次实现了人工手段模拟RFP[图4(B)].类似于对天然RFP生色团结构的限制,DNA G4为RFP生色团类似物的堆叠提供了理想的封装位点,并将它们限制在平面构象中以实现高效发射,激活红色和偏远光谱区域(λem=583~668 nm)的明亮荧光.与RFP相比,RFP的DNA模拟物具有明亮的红色发光、大的斯托克斯位移、抗光漂白和双光子荧光特性.我们进一步将该模拟物用于活细胞膜蛋白的标记,实现了膜蛋白的长时间示踪和双光子组织成像.
开发模拟天然荧光蛋白的合成材料,对于阐明荧光蛋白的发光机制和创造新的荧光响应工具和生物传感器至关重要.生色团骨架和核酸结构的可设计性在构建不同荧光蛋白的结构模拟物方面具有很大的潜力.同时,也需要进一步提高荧光蛋白结构模拟物的亮度和光稳定性,并开发更多的结构模拟物.
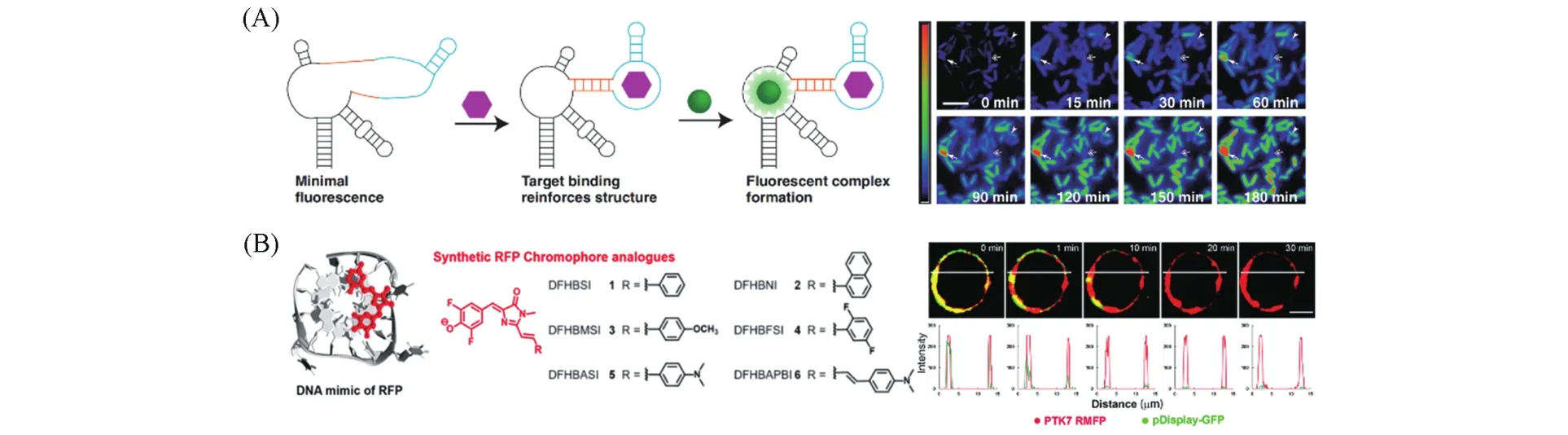
Fig.4 Representative applications of fluorescent protein mimics in biosensing
5 总结与展望
本文对荧光蛋白结构改造和模拟方法及其在生物传感中的代表性应用进行了阐述和分析.这些结构改造方法有效地优化了荧光蛋白的性质,发展了亮度更高、光稳定性、成熟更快以及不同颜色变体的荧光蛋白,赋予了荧光蛋白自组装、抗聚集等功能,扩展了荧光蛋白在生物传感、生物成像等领域的应用.在这些改造过程中,研究者对于荧光蛋白的结构与发光机制有了更好的了解,有助于开发性质更优且可用于深层组织和全身成像的荧光蛋白工具.通过对荧光蛋白的结构模拟,获得了人工手段模拟的荧光蛋白类似物,这是一个令人兴奋的概念,扩大了相关的检测目标范围.目前,仍然需要进一步改善荧光蛋白结构模拟物的亮度和光稳定性,以实现其在哺乳细胞内的有效成像.另外,基于对GFP的结构研究策略也为探究其它荧光蛋白在生物学领域中的进程提供了设计思路.
