TIR/BB-loop mimetic AS-1 protects vascular endothelial cells from injury induced by hypoxia/reoxygenation
2020-11-11ZhijiaZhangYuxingHouJiantaoLiChaoTangLinliQueQianTanYuehuaLi
Zhijia Zhang, Yuxing Hou, Jiantao Li, Chao Tang, Linli Que, Qian Tan, Yuehua Li,✉
1Department of Pathophysiology, Nanjing Medical University, Nanjing, Jiangsu 211166, China;2Department of Plasticsurgery, Drum Tower Hospital, Nanjing, Jiangsu 211100, China.
Abstract Morphological and functional abnormalities of vascular endothelial cells (VECs) are risk factors of ischemiareperfusion in skin flaps. Signaling pathway mediated by interleukin-1 receptor (IL-1R) is essential to hypoxia/reoxygenation (H/R) injury of VECs. While the TIR/BB-loop mimetic (AS-1) disrupts the interaction between IL-1R and myeloid differentiation primary-response protein 88 (MyD88), its role in the VECs dysfunction under H/R is unclear. In this study, we first showed that there was an infiltration of inflammatory cells and the apoptosis of VECs by using a skin flap section from patients who received flap transplantation. We then showed that the H/R treatment induced apoptosis and loss of cell migration of endothelial cell line H926 were attenuated by AS-1. Furthermore, our data suggested that AS-1 inhibits the interaction between IL-1R and MyD88, and subsequent phosphorylation of IκB and p38 pathway, as well as the nuclear localization of NF-KB subunit p65/p50. Thus, this study indicated that the protective role of AS-1 in H/R induced cellular injury may be due to the AS-1 mediated down-regulation of IL-1R signaling pathway.
Keywords: AS-1, vascular endothelial cells, hypoxia/reoxygenation, IL-1R, NF-κB, MAPK
Introduction
Flap transplantation is commonly used to repair tissue defect and reconstruct organs. The transplanted flaps (random, axial, or free) may undergo ischemia and reperfusion, a complex pathophysiological process of injury. Experimental studies have shown that oxygen free radical damage and calcium overload can cause vascular endothelial apoptosis,inflammatory response and other lesions, all contribute to ischemia-reperfusion injury[1-3].
Vascular endothelial cells participate in various physiological activities, such as synthesizing biological substances, contracting blood vessels,maneuvering blood flow, activating blood coagulation and regulating platelet aggregation[4-7]. In skin flaps,ischemia-reperfusion (I/R) can inflict endothelial cells with morphological and functional abnormalities, such as apoptosis, inflammatory substance synthesis[8-9].
As a key mediator in immune and inflammatory responses, IL-1 undergoes its signaling pathway through the activation of interleukin-1 receptor (IL-1R)[10-11]followed by nuclear factor-κB (NF-κB)signaling pathway, a crucial link between immune and inflammatory systems[12], whose function depends on myeloid differentiation primary-response protein 88(MyD88)[13]. The IL-1R signaling pathway is engaged in skin flap ischemia-reperfusion injury. TIR/BB-loop mimetic (AS-1) has a structural similarity to the TIR/BB domain of MyD88, and can inhibit the downstream signal transduction caused by IL-1[14].Our previous study has found AS-1 can relieve myocardial ischemia-reperfusion injury by inhibiting MyD88-dependent signaling pathway[15]. But whether AS-1 can decrease H/R-induced injury in vascular endothelial cells remains to be elucidated.
Here, by using paraffin sections made of skin flaps from patients along with HE and TUNEL staining, we found inflammation may play a critical role in endothelial cell I/R injury, and we examined the role of AS-1 in repressing endothelial cell injury induced by H/R. Furthermore, AS-1 improved VECs viability and attenuated macrophage migration, presumably by inhibiting the interaction between IL-1R and MyD88,and following phosphorylation of IκB and p38 pathway, as well as the nuclear localization of NF-KB subunit p65/p50.
Materials and methods
Cell culture and reagents
Human umbilical vein endothelial cells H926(ATCC, USA) were maintained in HG-DMEM medium (Life Technologies, USA) supplemented with 10% fetal bovine serum in a humidified atmosphere containing 5% CO2at 37 °C.
Cell treatment
AS-1 was dissolved in DMSO, and stored at-20 °C. For the cell viability test, H926 cells were stimulated with 0, 25, 50, and 100 μmol/L of AS-1 for 72 hours. To evaluate the protective effect of AS-1,H926 cells were pretreated with 50 μmol/L of AS-1 or DMSO for 30 minutes. H/R group cells were placed in a humidified atmosphere containing 1% O2and 5%CO2for 4 hours, and then 20% O2and 5% CO2for required time.
Tissue paraffin section and staining
The normal, necrotic, and border areas of the skin flap were fixed with 4% formalin. HE (Beyotime,China) and TUNEL (Roche, Germany) staining were performed according to the manufacturer's instructions. The skin flap tissue was obtained from Taixing People's Hospital, and the study was approved by Ethical Committee of Taixing People's Hospital.
Western blotting assay
Cytoplasmic proteins were prepared from harvested cells. Western blotting was performed as described previously[9]. Briefly, the cytoplasmic proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto PVDF membranes (Cell Signaling Technology, USA). The membranes were incubated with the appropriate primary antibodies followed by incubation with peroxidase-conjugated secondary antibodies. The signals were detected using the ECL system. The same membranes were probed with antibodies for GAPDH (1:5000, Sigma, USA). The signals were quantified by scanning densitometry. Computerassisted image analysis was performed using Image J.The primary antibodies included anti-cleaved caspase-3, anti-Bax, anti-Bcl-2, anti-phospho-p38, antiphospho-IκB, anti-Tublin, and anti-LaminB (all diluted in 1:1000, Cell Signaling Technology).
Flow cytometric analysis
The assay was performed according to the manufacturer's instruction. Briefly, both treated and untreated cells were harvested, washed with PBS,suspended in Annexin V binding buffer (10 mmol/L HEPES, pH 7.4; 2.5 mmol/L CaCl2, 140 mmol/L NaCl), stained with Annexin V-FITC (BD Biosciences, USA), and evaluated by the FACScalibur flow cytometer.
Cell viability assay by CCK8
H926 cells were seeded in a 96-well plate (5 000 cells/well), pre-incubated for 24 hours in a humidified incubator (37 °C, 5% CO2), and treated as above.Then after 10 μL of CCK-8 solution was added into each well, the plate was incubated for 4 hours. The absorbance was measured using a microplate reader at 450 nm.
Scratch wound closure assay
H926 cells were seeded in six-well plates and allowed to proliferate to a density of 80% (the cells nearly attaching to each other). Then the cell monolayer was scratched using a 1 mL pipette tip.Each well was washed once with growth media to remove cell debris. To record scratch wound closure,the images were captured at 0, 12, 24, and 48 hours until the wound was closed.
Transwell assay
RAW264.7 cell solution was plated on top of the filter membrane in a transwell insert and incubated at 37 °C and 5% CO2. H926 cells were plated on the bottom of the lower chamber in a 24-well plate and treated as described above. The transwell insert was added without generating bubbles, incubated for 24 hours, and then removed from the plate. A cottontipped applicator was used to carefully remove the media. The cells that had not migrated from the membrane top were retained. Then, 70% ethanol was added into a 24-well plate to fix the cells. After removing the transwell insert, the remaining ethanol was removed from the top of the membrane using a cotton-tipped applicator. The membrane was positioned into a 24-well plate with 0.2% crystal violet and incubated at room temperature for 5-10 minutes for staining. The crystal violet was gently removed from the membrane top with a pipette tip.The cells on the membrane were photographed with SLR camera.
Total RNA extraction and qRT-PCR
Total RNA was extracted from cells using RNAiso reagent (Takara Biotechnology, Japan) and RT-PCR assays were performed with the Prime-ScriptTMRT detection kit (Takara Biotechnology) as previously described. Amplification and detection of specific products were performed using the ABI Prism 7500 sequence detection system with the cycles indicated by the Prime-ScriptTMRT detection kit. As an internal control, HPRT primers were used for RNA template normalization. qRT-PCR primers were presented in Table 1. Relative levels of the target gene mRNA expression were calculated and expressed as 2-△△Ct.

Table 1 PCR primer sequences
Co-immunoprecipitation
Cytoplasmic proteins were incubated with anti-IL-1R antibody (Santa Cruz Biotechnology, USA) for 2 hours at 4 °C on a rotator. Then, 20 μL protein A/G beads (Santa Cruz Biotechnology) was added to each sample followed by incubation overnight at 4 °C on a rotator. The samples were centrifuged briefly in a microcentrifuge and washed three times in the 1 mL lysis wash buffer (250 mmol/L NaCl, 1% NP-40, 1 mmol/L PMSF, 5 mg/mL aprotinin, and 5 mg/mL leupeptin) (pH 8.0). Loading buffer (pH 6.8) was added to each sample and boiled for 5 minutes.
Statistical analysis
All results were expressed as mean±standard deviation (SD). One-way ANOVA was used to test the differences between the groups. Comparisons between two groups were performed using Student's ttest. A P-value <0.05 was considered statistically significant.
Results
Ischemia-reperfusion of skin flap underwent pathological changes
Skin flap samples were obtained from patients who suffered from burns and received flap transplantation.The normal, necrotic, and border samples were taken in situ. The border samples showed marked edema and inflammatory cell infiltration; the necrotic samples presented more inflammatory cells with disappeared organizational structure (Fig. 1A).TUNEL staining indicated a larger number of apoptotic cells in necrotic tissues than in the normal tissues (Fig. 1B).
AS-1 ameliorated the viability of vascular endothelial cell injured by H/R
To verify the effect of AS-1 in flap ischemiareperfusion injury, we employed vascular endothelial cells in vitro. Firstly, we tested the cytotoxicity AS-1 exerted to vascular endothelial cells. The vascular endothelial cells were stimulated by AS-1 at different concentrations for 72 hours. Cell viability was detected by the CCK8 kit. There was no significant toxicity in endothelial cells was detected with different concentrations of AS-1 (Supplementary Fig. 1,available online). Further, the viability of vascular endothelial cells was significantly inhibited by H/R compared with the control group. However, AS-1 could significantly ameliorate the H/R-impaired cellular viability, while the solvent control group showed no difference (Fig. 2A and B).
AS-1 attenuated H/R-induced vascular endothelial cell apoptosis
FACS analysis indicated H/R stimulation significantly promoted the apoptosis of vascular endothelial cells compared with the control group. In contrast, AS-1 could significantly prevent cell apoptosis compared to the solvent control (Fig. 3A).H/R stimulation significantly increased the expression of apoptotic protein Bax, decreased the expression of anti-apoptotic protein Bcl-2, while AS-1 prevented these H/R-induced changes (Fig. 3B). Also, AS-1 decreased the activation of caspase-3 as indicated by cleaved-caspase-3 (Fig. 3C).
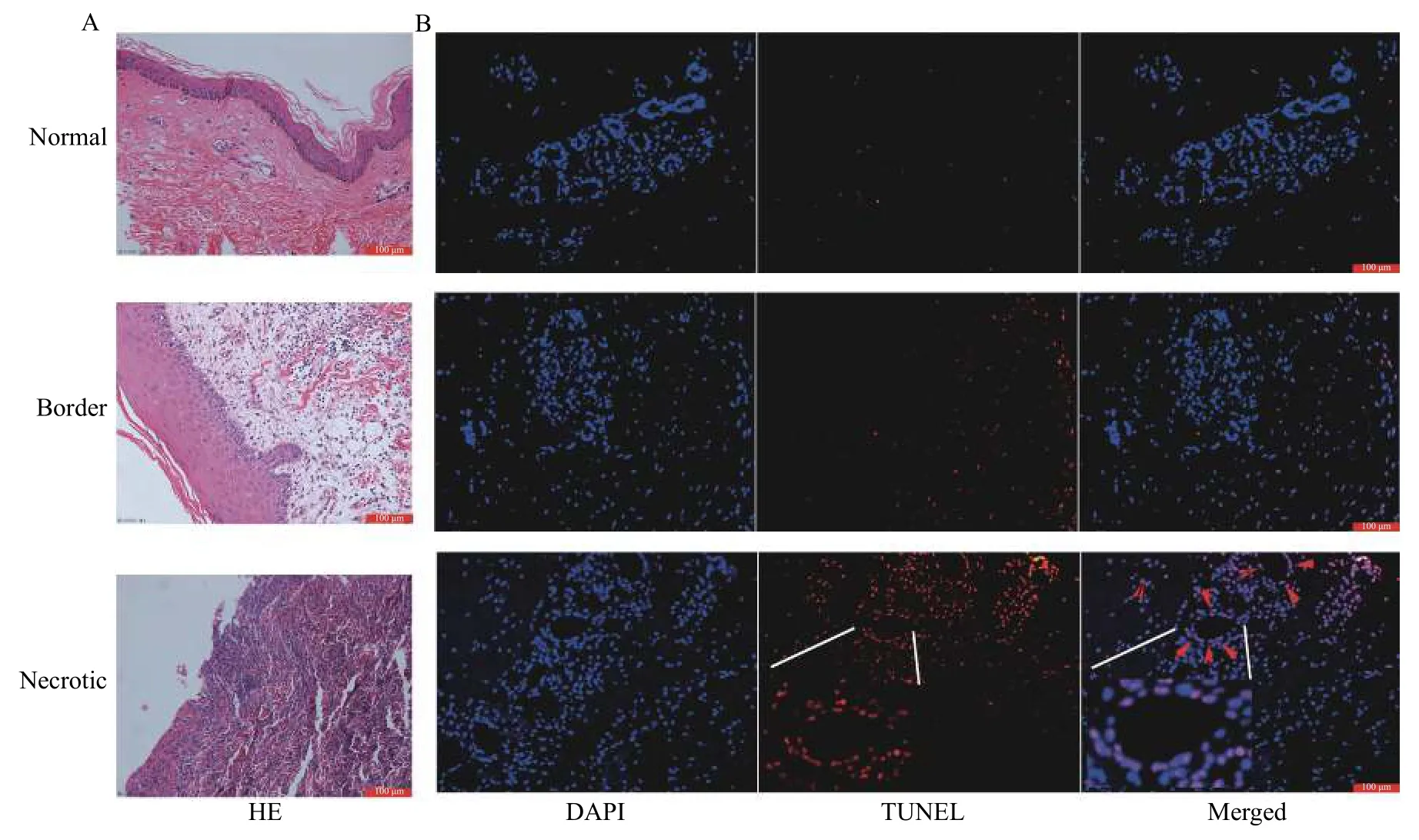
Fig. 1 Different flap morphology after flap transplantation. A: HE staining, more inflammatory cell infiltration in the perivascular and subcutaneous tissues at the edge of the necrotic flap; the disordered subcutaneous tissue of the necrotic flap, a large number of infiltrated inflammatory cells, significantly reduced vascular structure. B: TUNEL staining, obvious cell apoptosis, including vascular endothelial cell apoptosis, in the subcutaneous tissue of the necrotic flaps. Scale bar, 100 μm.

Fig. 2 AS-1 ameliorated viability of vascular endothelial cells impaired by H/R. A: H926 cells seeded in 96-well plates and pretreated with AS-1 or control solvent for half an hour before being subjected to hypoxia for 4 hours and reoxygenated for 1 hour;cell viability meas-ured by CCK8 kit. B: Cell pictures captured under Carl Zeiss digital microscope. n=3,*P<0.05 vs. Control; #P<0.05 vs. H/R+DMSO. Scale bar, 200 μm.
AS-1 promoted the repair in H/R-damaged vascular endothelial cells
H/R stimulation significantly inhibited the repair of endothelial cells, as evidenced by their prolonged time spent in repairing. AS-1 pretreatment significantly promoted the repair of vascular endothelial cells, but this change was not observed in the solvent control group (Fig. 4).
AS-1 inhibited H/R-activated and IL-1R mediated signaling pathway
The interaction between IL-1R and MyD88 through TIR/BB domain is essential to signaling transduction.Co-IP indicated H/R treatment enhanced the interaction between IL-1R and MyD88, while AS-1 repressed this process (Fig. 5A). Compared with the control group, H/R obviously induced the phosphorylation of p38 and IκB protein. However,after AS-1 pretreatment, the expression of p-p38 protein was significantly inhibited by H/R, which was found in the solvent control group (Fig. 5B and C).Immunofluorescence indicated that p50 subunit assembled in the nucleus. WB indicated that p65 subunit mainly appeared in the nuclear under H/R, but AS-1 reversed the above effect (Fig. 5D and E).
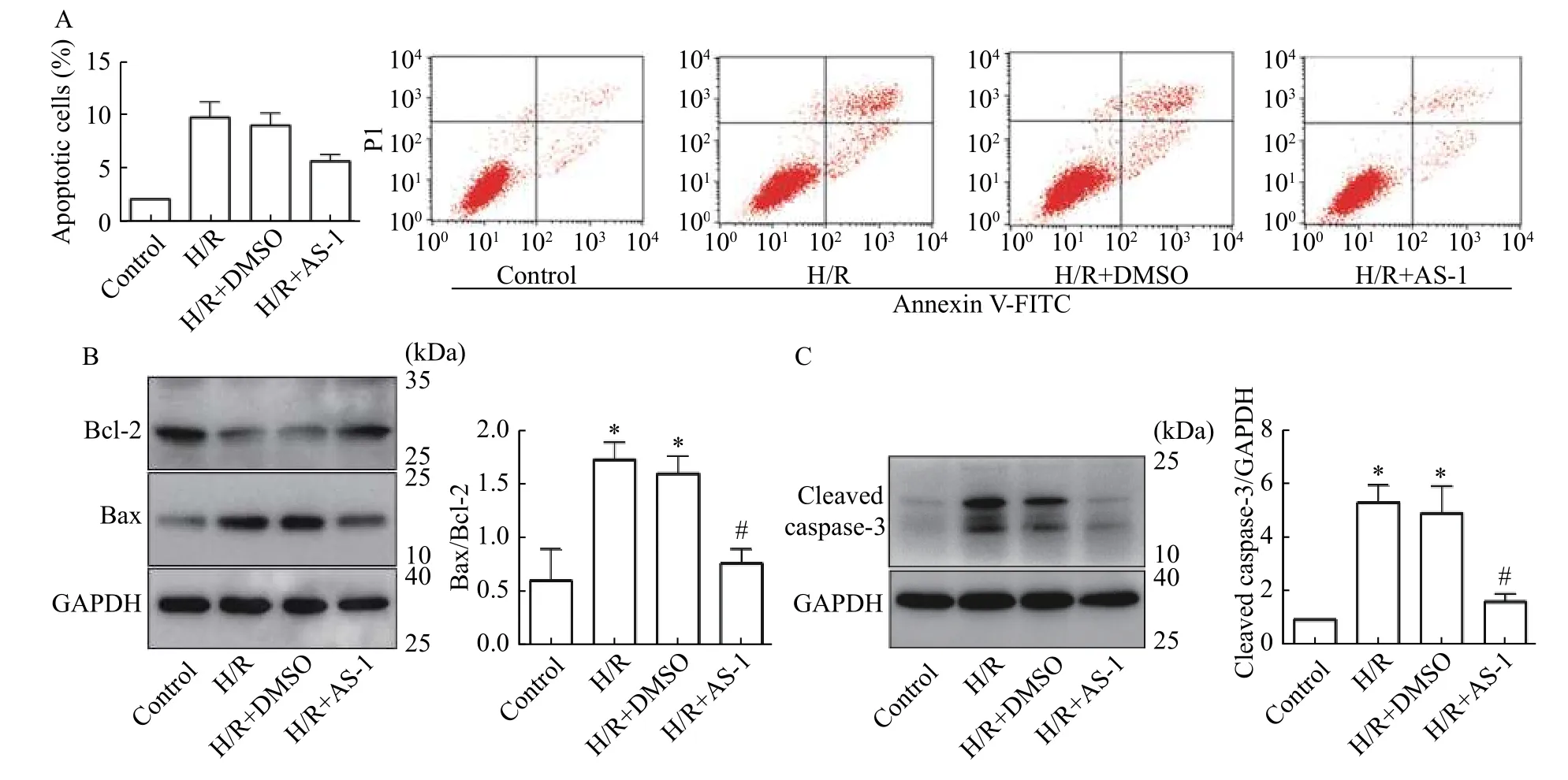
Fig. 3 AS-1 attenuated H/R-induced vascular endothelial cell apoptosis. A: Vascular endothelial cells were treated as described. Flow cytometry was used to detect apoptosis. B and C: Western blotting was performed to detect molecular involved in cell apoptosis. n=3,*P<0.05 vs. Control; #P<0.05 vs. H/R+DMSO.

Fig. 4 AS-1 ameliorated cell migration which was impaired by H/R. H926 cells were seeded in 96-well plates and pretreated with AS-1 and control solvent for half an hour before being subjected to hypoxia for 4 hours. The scratch wound closure assay was performed, images were captured at 0, 12, 24, and 48 hours.
AS-1 ameliorated macrophage migration induced by conditioned medium from H/R-treated cells
H/R treatment significantly promoted the synthesis of inflammatory cytokines (IL-1, TNF-α, MCP-1, IL-6 and IL-8) in vascular endothelial cells compared with the control group, however, the pretreatment of AS-1 before hypoxia significantly inhibited the expression of inflammatory cytokines, which was not found in the solvent control (Fig. 6A-E). Further, the conditioned medium from H/R treatment group significantly promoted the migration of macrophages.By contrast, no similar effect was detected in the conditioned medium derived from the AS-1 pretreated group (Fig. 6F).

Fig. 5 AS-1 inhibited the activation of IL-1R mediated signaling pathway. A: Co-immunoprecipitation was taken using anti-IL-1R antibody to analyze the interaction between IL-1R and MyD88. B and C: Cells total lysate was collected to perform Western blotting assays. D:Immunofluorescence and confocal microscopy were employed to detect the p50 nuclear localization. E: Subcellular fractionation was collected to detect p65 location, GAPDH was taken as the cell plasma protein marker and Laminin B was taken as the nuclear protein marker.Scale bar, 100 μm.
Discussion
In the present study, cell apoptosis and inflammatory response were found in ischemiareperfusion-injured skin flap tissue. In this study, we used AS-1 to ameliorate H/R-induced endothelial cell injury. Indeed, in the case of hypoxia and reoxygenation, the viability of the vascular endothelial cells was significantly decreased, cell apoptosis significantly increased, and the cell migration significantly slowed down; AS-1 efficiently protected the cells through attenuating the phosphorylation of p38 and IκB. H/R stimulation significantly induced synthesis of inflammatory cytokines IL-1, TNF-α and chemokine MCP-1 in vascular endothelial cells, but this induction was inhibited by AS-1. AS-1 also ameliorated macrophage migration induced by conditioned medium from H/R treated vascular endothelial cells.
As mentioned before, ischemia reperfusion is a powerful risk factor for skin flap injury[1-3], so finding the exact mechanism and new treating strategy is urgently needed. The ischemia-reperfusion flap injury features abnormal morphology and function of endothelial cells[8-9]. We found vascular endothelial cell damage was significantly aggravated after H/R treatmentin vitro, mainly due to the decreased cell viability and increased apoptosis, which is similar to the findings of previous studies[16].
During ischemia-reperfusion injury, MAPKs[17-18]and NF-κB[19]are activated in damaged vascular endothelial cells. MAPKs are intracellular serine/threonine protein kinases, including ERK, JNK,and p38MAPK. Recent studies have shown that ERK,JNK, and p38MAPK are closely related to acute inflammation and skin cancer[20]. In ischemiareperfusion flaps, the MAPKs are also activated[21].Ischemia-reperfusion can also activate NF-κB signaling pathway. NF-κB, an important nuclear transcription factor, is indispensable for immune inflammation. Studies have shown that ischemiareperfusion can induce the production of cytokines,such as IL-1β, TNF-α in endothelial and inflammatory cells. Notably, the expression of inflammatory mediators and factors (such as IL-1, IL-6 and TNF-α)exacerbates the inflammatory response, while the inhibition of NF-κB expression prevents the release of inflammatory factors[22-23]. It is suggested that the activation of inflammatory signaling pathway participates in ischemia-reperfusion-induced injury of vascular endothelial cells.

Fig. 6 AS-1 repressed the inflammatory cytokine synthesis induced by H/R, and repressed the migration of macrophages induced by conditional medium from H/R treated cells. A-E: Total mRNA was extracted and qRT-PCR was performed to detect expression levels of IL-1, TNF-α, MCP-1, IL-6, and IL-8. F: RAW264.7 macrophages were seeded in transwell chambers. After H/R stimulation, the transwell chambers were inserted into a 12-well plate and crystal violet staining was performed 24 hours later. n=3, *P<0.05 vs. Control; #P<0.05 vs.H/R+DMSO.
AS-1 is a lipids-soluble substance that easily enters the cell and mimics the sequence of a tripeptide[(F/Y)-(V/L/I)-(P/G)] in the MyD88-TIR-BB loop.AS-1 can bind to MyD88-TIR region competing with IL-1R-TIR domain and inhibit IL-1β-increased phosphorylation of IRAK-1 and p38-MAPKin vitro.In this study, we found that AS-1 could significantly inhibit the phosphorylation of p38 and IκB. In conclusion, TIR/BB-loop analog AS-1 can protect vascular endothelial cells from H/R injury through inhibiting the expression of inflammatory cytokines induced by H/R. This mechanism involves the activation of MAPKs and NF-κB ensuing IL-1R down-regulation. The present study provides a theoretical basis for clinical prevention and treatment of flap ischemia-reperfusion injury.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81470418 and No.81770230).
杂志排行

THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Pre-evaluation of humoral immune response of Bactrian camels by the quantification of Th2 cytokines using real-time PCR
- Comparison of the modified Wiltse's approach with spinal minimally invasive system and traditional approach for the therapy of thoracolumbar fracture
- Exposure to environmental bisphenol A inhibits HTR-8/SVneo cell migration and invasion
- Cumulative live birth rates of in vitro fertilization/intracytoplasmic sperm injection after multiple complete cycles in China
- Cofilin participates in regulating alpha-epithelial sodium channel by interaction with 14-3-3 isoforms
- Postprandial dyslipidemia in insulin resistant states in adolescent populations