依非韦伦中间体的不对称合成
2020-10-30张方方周毅博吴广文
李 灿,张方方,周毅博,王 波,祝 航,吴广文
新型反应器与绿色化学工艺湖北省重点实验室(武汉工程大学),湖北 武汉 430205
依非韦伦(Efavirenz),化学名称为:(S)-(-)-6-氯-4-环丙基乙炔基-1,4-二氢-4-三氟甲基-2H-3,1-苯并噁嗪-2-酮[1-2],是由默沙东公司研发的一种非核苷类逆转录酶抑制剂,于1998年经美国FDA 批准上市。临床上,依非韦伦是抗HIV 感染“鸡尾酒疗法”的重要组成部分,该组合包括依非韦伦、齐多夫定和拉米夫定,该法疗效佳、半衰期长、耐受性好,能迅速控制病毒[3-5]。由于其具有巨大的市场前景,所以优化依非韦伦的合成工艺具有重要意义。
(S)-1-(2-氨基-5-氯苯基)-1-三氟甲基-3-环丙基-2-丙炔-1-醇是合成依非韦伦的关键中间体,目前其合成方法已有多篇文献报道[6-8]。1995年,默克公司[9-10]首次报道了依非韦伦中间体的不对称合成方法,加成反应几分钟之内得到ee 值达98%的手性叔醇。但该法中所用正丁基锂为危险试剂使用条件苛刻,且该反应需在低温(-50 ℃)进行,能耗较高,所用配体价格昂贵,反应中需频繁进行氨基保护和脱保护,操作繁琐,步骤较多,不利于工业生产。1999年,Tan 和其同事[11]开发了一种锌介导的,4-氯-2-(三氟乙酰基)苯胺与环丙乙炔格氏试剂不对称合成手性中间体的方法,该法通过格氏试剂和非手性试剂的添加降低了反应物的用量,提高了收率和光学纯度。胡争朋等[12-13]采用了自催化的方法,反应体系中加入少量的纯品手性中间体,大大减少了手性配体的用量,但该法收率和光学活性较低,收率为85.6%,ee 值85.6%,总收率为69.5%,难以达到工业化生成要求[14]。因此,寻找一条操作简单,成本低廉且适合工业化生产的合成路线显得尤为重要,本文报道了一条依非韦伦手性中间体新的合成路线(见图1)。
该合成路线用(Boc)2O 对对氯苯胺(1)的氨基进行保护,所得中间体N-叔丁氧基羰基-4-氯苯胺,低温下与三氟乙酸乙酯在三氯化铝的催化下发生傅-克酰基化反应,产物在氯化氢/乙酸乙酯体系中脱保护,经饱和碳酸氢钠碱化得4-氯-2-(三氟乙酰基)苯胺,在较温和的条件下与环丙乙炔氯化镁发生不对称加成反应得到(S)-1-(2-氨基-5-氯苯基)-1-三氟甲基-3-环丙基-2-丙炔-1-醇,核磁共振氢谱及核磁共振碳谱对化合物进行了分析和表征,高效液相色谱(high performance liquid chromatography,HPLC)检测对映体过量(ee 值)达98.2%,总收率达79.8%。此路线操作简便,成本低廉,易于工业化放大生产。

图1 (S)-1-(2-氨基-5-氯苯基)-1-三氟甲基-3-环丙基-2-丙炔-1-醇的合成Fig.1 Synthetic route of(S)-1-(2-amino-5-chlorophenyl)-1-trifluoromethyl-3-cyclopropyl-2-propyne-1-ol
1 实验部分
1.1 试剂与仪器
试剂:对氯苯胺,手性配体(1R,2S)-1-苯基-2-(吡咯烷基)-1-丙醇(分析纯,浙江沙星医药化工有限公司);四氢呋喃(tetrahydrofuran,THF),2-甲基四氢呋喃(2-methyltetrahydrofuran,2-MeTHF);二碳酸二叔丁酯[di-tert-butyl dicarbonate,(Boc)2O];二氯甲烷(methylene chloride,DCM);氯化铝(aluminum chloride,Al3Cl);乙酸乙酯(ethyl acetate,EA);三乙胺(triethylamine,Et3N);碳酸氢钠(sodium bicarbonate,NaHCO3);甲苯(toluene);氢化钠(sodium hydride,NaH);氯化锌(zinc chloride,ZnCl2);三氟乙酸乙酯(ethyl trifluoroacetate,TFAET)等(分析纯,国药集团有限公司)。
仪器:Agilent-400MHz 型核磁共振波谱仪(CDCl3和DMSO-d6为溶剂,TMS 为内标);DIONEX Ultimate 3000 型高效液相色谱仪;RY-1 熔点仪(天津市天分分析仪器厂)。
1.2 HPLC 检测条件
1.2.1 纯度的检测方法 HPLC 归一化法:色谱柱,Inertsil ODS-3(250 mm×4.6 mm×5 μm);流动相,V(乙腈)∶V(水)=65∶35;检测波长252 nm;柱温30 ℃;流速1 mL/min。
1.2.2 ee 值的检测方法 HPLC 归一化法:色谱柱,大赛璐AD-H 柱(4.6 mm×250 mm×5 μm);流动相,V(正己烷)∶V(异丙醇)∶V(三乙胺)=850∶150∶1;检测波长250 nm;柱温30 ℃;流速1 mL/min[2]。
1.3 实验方法
1.3.1 N-叔丁氧基羰基-4-氯苯胺(2)的合成 将对氯苯胺(29.32 g,0.23 mol)以及Et3N(26.3 g,0.26 mol)加入到THF(150 mL)和水(150 mL)的混合物中,搅拌直至化合物全部溶解,在40 min 内向所得的浆液中滴加(Boc)2O(54 mL,0.26 mol);滴毕,在30 ℃下搅拌反应6 h 后停止。反应混合物用乙酸乙酯溶解,分别用水和饱和食盐水溶液洗涤,于55 ℃减压浓缩至干,刮出,干燥1 h,得白色固体50.64 g,收率为96.7%,m.p.103~105 ℃。1H NMR(400 MHz,CDCl3)δ:7.85(d,J=8.8 Hz,2H),7.34(d,J=8.4 Hz,2H),6.65(s,1H),1.53(s,9H);13C NMR (100 MHz,CDCl3)d:176.62,136.66,129.15,128.84,121.39,89.62,27.63。
1.3.2 4-氯-2-(三氟乙酰基)苯胺盐酸盐(3)的合成 将二氯甲烷(350 mL)降温至-20 ℃,加入无水三氯化铝(58.76 g,0.44 mol),搅拌均匀,在1 h 内向混合液中滴加三氟乙酸乙酯(35.52 g,0.25 mol),保持温度-23~-20 ℃,反应2 h 后。降温至-25 ℃,30 min 内分3 批加入N-叔丁氧基羰基-4-氯苯胺(47.82 g,0.21 mol)。升温至25 ℃,保温搅拌3 h。将反应液缓慢加至冰水(350 mL)中,控制温度<15 ℃,滴加完毕后,于室温下搅拌反应1 h。将上述反应液用乙酸乙酯溶解,用蒸馏水洗涤,0 ℃下向有机相中通入氯化氢(0.42 mol)气体,通入完毕后,自然升温至30 ℃搅拌4 h,将反应液降温至0~5 ℃并保温2 h,有白色固体析出,过滤收集产物,滤饼用乙酸乙酯淋洗,于50 ℃真空干燥10 h 得白色固体47.12 g,收率为86.3%,m.p. 159~162 ℃。
1H NMR(400 MHz,DMSO-d6) δ:7.47(d,J=7.1 Hz,2H),7.21(s,1H),6.99(d,J=9.8 Hz,3H)。
1.3.3 4-氯-2-(三氟乙酰基)苯胺(4)的合成 向上一步得到的化合物3(45.00 g,0.173 mol)和乙酸乙酯(350 mL)中加入饱和NaHCO3溶液(230 mL,0.9 mol/L),混合物调至pH=7~8,25 ℃下搅拌反应1 h 后。分液,有机相用蒸馏水洗涤,减压浓缩至干,刮出,在45 ℃下真空干燥4 h,得淡黄色粉末36.48 g,收率为99.3%,m.p. 97~98 ℃。1H NMR(400 MHz,CDCl3)δ:7.70(s,1H),7.32(dd,J=9.0,2.2 Hz,1H),6.69(d,J=9.0 Hz,1H),6.46(s,2H);13C NMR(100 MHz,CDCl3)δ:176.62,136.66,129.15,128.88,121.39,39.62,27.63。
1.3.1 (S)-1-(2-氨基-5-氯苯基)-1-三氟甲基-3-环丙基-2-丙炔-1-醇(5)的合成 将镁屑(3.30 g,0.14 mol)加至2-MeTHF(25 mL)中,在氮气保护下搅拌反应,并滴加溴乙烷(1 mL),体系温度升至41 ℃时,有少量气体产生,溶液由无色变为灰色;设置水浴温度30 ℃,并开始滴加1-氯丁烷(12.79 g,0.13 mol)与2-MeTHF(40 mL)的混合液,滴加完毕后,升温至50 ℃,搅拌反应3 h。降温至10~15 ℃,缓慢滴加环丙乙炔(8.89 g,0.13 mol),滴毕,10~20 ℃保温,持续搅拌2 h,制得环丙乙炔氯化镁,放置备用。
在氮气保护下,将氢化钠(60%,13.00 g,0.325 mol)加至2-MeTHF(40 mL)中搅拌反应,降温至10~15 ℃,缓慢滴加新戊醇(9.12 g,0.103 mol)、手性配体(1R,2S)-1-苯基-2-(吡咯烷基)-1-丙醇(25.80 g,0.126 mol)和甲苯(120 mL)的混合液,滴毕,控制温度<25 ℃,搅拌反应2 h。保温结束,降温至-15 ℃,分2 批加入无水氯化锌(17.66 g,0.13 mol),加完后升温至25~30 ℃,保温搅拌3 h。降温至10~15 ℃,滴加上述制备的环丙乙炔氯化镁,滴毕,在此温度保温搅拌3 h。降温至-20 ℃,一次性加入化合物4(24.22 g,0.108 mol),在-10 ℃下保温反应15 h。将反应液加至柠檬酸(1 mol/L,200 mL)中淬灭反应。分液,水层转移至回收瓶以回收有机配体,将有机层用200 mL 水洗涤,并将有机相浓缩至80 mL,加入甲苯(70 mL)再次浓缩至80 mL 以除去所有2-MeTHF。缓慢加入正庚烷(150 mL)。将混合物冷却至0 ℃,过滤收集固体,用正庚烷(30 mL)洗涤,干燥,得到白色固体30.13 g,收率为96.4%,纯度为98.6%,ee 值为98.2%,m.p.139~141 ℃。1H NMR(400 MHz,CDCl3)δ:7.49(d,J=2.0 Hz,1H),7.09(dd,J=8.6,2.4 Hz,1H),6.58(d,J=8.6 Hz,1H),4.55(s,3H),1.42~1.33(m,1H),0.96-0.73(m,4H);13C NMR(100 MHz,CDCl3)δ:143.97,130.98,130.67,126.09,124.40,123.24,121.28,94.22,75.26,71.19,9.16,9.11,0.00。
2 结果与讨论
2.1 (S)-1-(2-氨基-5-氯苯基)-1-三氟甲基-3-环丙基-2-丙炔-1-醇的合成
2.1.1 溶剂和底物浓度的筛选 该反应中格氏试剂的制备及手性中间体的不对称合成都需要在无水无氧条件下进行,对溶剂的除水要求较高。THF 易吸水,回收成本较高;2-MeTHF 为绿色工业溶剂,难与水互溶,易回收套用[7]。底物4-氯-2-(三氟乙酰基)苯胺的浓度对收率和ee 值影响较大。本研究对溶剂和底物的浓度进行了筛选,结果如表1 所示。
从反应结果可以看出,底物浓度相同时,两种溶剂体系的反应收率和ee 值相差不大,但2-MeTHF 比THF 更易回收套用,更符合绿色化学理念。底物的浓度对反应完成时间的影响较大。浓度过低,反应较慢,反应时间延长,产率和ee 值降低;浓度过高,又会发生反应物的聚集,降低反应收率和ee 值。综合考虑,以2-MeTHF/toluene 作为溶剂体系,底物浓度为0.45 mol/L。

表1 溶剂和底物浓度的筛选Tab.1 Screening of substrate concentrations and solvents
2.1.2 反应物配比对手性中间体合成的影响 本研究采用不对称合成的方法,以(1R,2S)-1-苯基-2-(吡咯烷基)-1-丙醇为手性配体,手性配体与新戊醇、氯化锌、环丙乙炔氯化镁形成金属配合物,从而诱导目标产物的生成。手性配体和氯化锌的多少对反应的收率和立体选择性有直接的影响[15-16]。本研究对反应物的摩尔配比进行考察,结果如表2 所示。
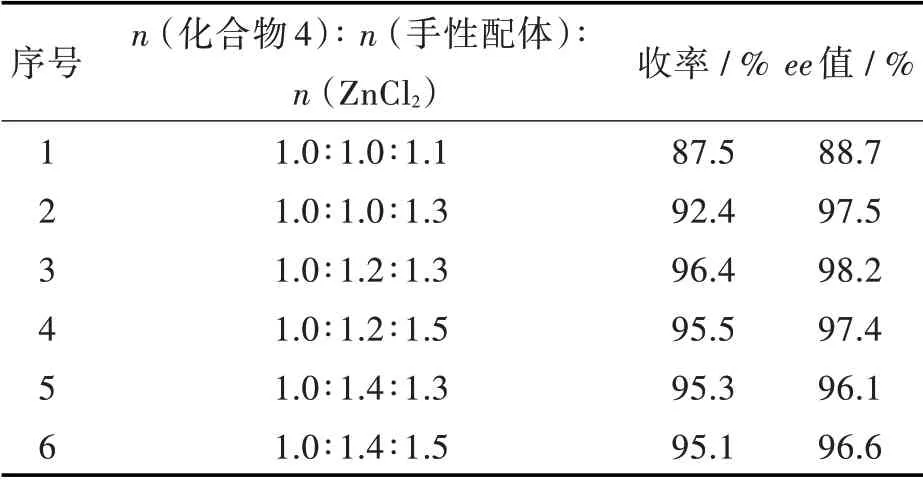
表2 反应物的摩尔配比对手性中间体合成的影响Tab.2 Effect of mole ratio of reactants on synthesis of chiral intermediate
从表2 可以看出,4-氯-2-三氟乙酰基苯胺(4)、手性配体与ZnCl2的摩尔配比为1.0∶1.2∶1.3 时,收率最高为96.4%,ee 值为98.2%,与文献[9]报道相近。用ZnCl2代替文献中危险且昂贵的Et2Zn,优化反应条件,降低了手性配体的用量,节约了成本,更加有利于工业化生成。由第2 组、第3 组和第5组实验结果可知,ZnCl2用量相同的情况下,手性配体与化合物4 的配比为1.2∶1.0 时收率和ee 值最佳,降低配比反应不完全,收率降低,配比超过1.2∶1.0 时收率和ee 值无明显变化。配体用量相同的情况下,由第3 组和第4 组实验结果可知,ZnCl2与化合物4 的配比为1.3∶1.0 时最佳,超过该配比,收率和ee 值无明显变化。综上,化合物4、手性配体与ZnCl2的摩尔配比为1.0∶1.2∶1.3 时,收率和ee 值最佳。
3 结 论
本文以对氯苯胺为起始原料,经氨基保护、酰化、脱保护、碱化、加成,不对称合成了(S)-1-(2-氨基-5-氯苯基)-1-三氟甲基-3-环丙基-2-丙炔-1-醇。优化了不对称合成手性中间体的反应条件,得出最佳工艺条件为:不对称加成反应中,溶剂体系选用2-MeTHF/toluene,底物浓度0.45 mol/L,物料配比为n(4-氯-2-三氟乙酰基苯胺)∶n(手性配体)∶n(ZnCl2)=1.0∶1.2∶1.3,收率最高为96.4%,ee 值为98.2%。该反应用廉价易得的ZnCl2代替危险且昂贵的Et2Zn,使用绿色工业溶剂2-MeTHF 代替THF,降低了手性配体的用量,纯化简单,溶剂回收率高,节约成本。该合成路线具有操作简单、反应条件温和、立体选择性较高的优点,适合工业化生产。
