小球藻多糖的分离纯化及理化性质
2020-10-28史瑞琴梁静静李大伟郭书贤马艳莉
史瑞琴,梁静静,李大伟,王 颉,郭书贤,马艳莉,
(1.南阳理工学院 河南省工业微生物资源与发酵技术重点实验室,河南 南阳 473004;2.河北农业大学食品科技学院,河北 保定 071000)
小球藻(Chlorella)是绿球藻目中的常见植物,属绿藻门,小球藻属,又称绿藻,它是地球上最早的生命形式之一,它是日本、中国等亚洲国家的传统食品[1]。多糖是小球藻中重要的营养物质,占小球藻粉的10%~20%,具有抗衰老、抗氧化及增强免疫等生物活性功能[2-5]。目前,市场上除小球藻片剂、胶囊等早期产品外,许多研究者还对小球藻饮料、面包、饼干等产品进行了深入研究[6-7],但对小球藻多糖类食品及多糖的理化特性鲜有报道。
近年来,已有研究证实多糖的结构与理化特性均会影响其在食品中的活性表征[8]。另外,在多糖类食品的加工过程中,为了降低加工能耗、增加产品口感,可通过更改施加的剪切应力调整溶液黏性[9]。天然多糖独特的流变学特性的研究可以预测其凝胶化或增稠性质,这不仅有助于食品的制造、分配、贮存和消费,而且有益于天然的凝胶剂、增稠剂或乳化剂的研发[10]。因此,为了提高多糖产品的种类及应用,减少制作过程中的能量消耗,需要对小球藻多糖的理化性质进行初步研究。
为了进一步探索小球藻多糖的应用价值,本实验首先通过酶法提取小球藻粉中的粗多糖,探讨了不同脱蛋白方法对小球藻多糖的影响,确定离子交换柱层析获得的纯化多糖中是否含有大量蛋白以及是否为均一性组分。同时研究不同纯化多糖的微观形态、单糖组成、流变学等特性,这对于扩大小球藻在食品领域中的应用具有重要的意义及参考价值。
1 材料与方法
1.1 材料与试剂
小球藻粉 西安仁邦生物科技有限公司;三氯乙酸(trichloroacetic acid,TCA) 天津市福晨化学试剂厂;氯仿、正丁醇 天津市科密欧化学试剂有限公司;纤维素酶(酶活力≥3 000 U/g)、木瓜蛋白酶(酶活力≥60 000 U/g)、牛血清、1-苯基-3-甲基-5-吡唑啉酮(1-phenyl-3-methyl-5-pyrazolone,PMP)、DEAESepharose Fast Flow、Sephadex G-200填料、单糖标准品北京索莱宝科技有限公司。
1.2 仪器与设备
H.SWX-600BS恒温水温箱 常州朗博仪器制造有限公司;Ne-ofuge 15R冷冻离心机 上海力申科学仪器公司;SHB-III循环水式多用真空泵 上海天星科学仪器有限公司;BT300-2J冻干机 新阳设备制造有限公司;UV-1700PC紫外-可见分光光度计 上海美析仪器有限公司;S3400N扫描电子显微镜(scanning electron microscope,SEM) 日本Hitachi公司;Breeze高效液相色谱(high performance liquid chromatography,HPLC)仪 美国Waters公司;MCR302高级旋转流变仪奥地利安东帕股份有限公司。
1.3 方法
1.3.1 小球藻多糖的提取
将小球藻粉与蒸馏水按照质量比1∶20混匀,超声30 min后添加1.45%复合酶(纤维素与木瓜蛋白酶的质量比为1∶1)并调pH 5,酶解30 min后灭酶。使溶液置于70 ℃浸提4 h,4 500 r/min离心15 min,取上清液与3 倍体积乙醇混合并于4 ℃过夜,离心收集醇沉物,复溶后浓缩至原体积的1/2,以便除去样品中的乙醇,此时粗多糖的提取率可达(6.56±0.52)%。
1.3.2 小球藻多糖的分离纯化与鉴定
1.3.2.1 不同脱蛋白方法的比较
TCA法脱蛋白:取10 mL浓缩液置于试管中,添加TCA使其质量分数分别达到1%、2%、3%、4%、5%、6%,4 ℃过夜后于5 000 r/min离心15 min,考察TCA质量分数对溶液中蛋白质脱除率与多糖损失率的影响[11]。
Sevag法脱蛋白:向浓缩液中加入1/2体积的Sevag试剂(氯仿-正丁醇(4∶1,V/V)),剧烈振荡20 min,移入分液漏斗中静置20 min,弃去中间与下层物质,多次重复上述工艺至观察到无明显的中间蛋白层[12],研究脱除蛋白次数对溶液中蛋白质脱除率与多糖损失率的影响。计算如式(1)、(2)所示:
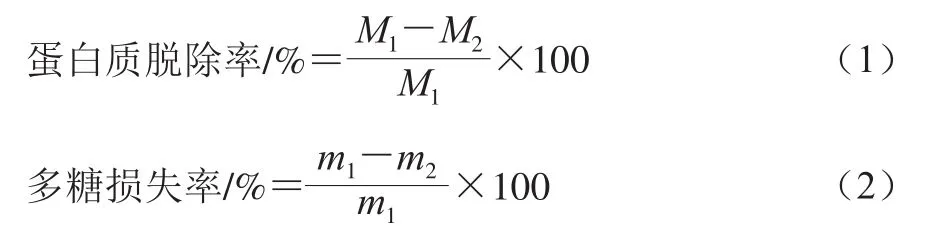
式中:M1、M2分别为脱蛋白前、后溶液中蛋白质含量;m1、m2分别为脱蛋白前、后溶液中多糖含量。
1.3.2.2 除杂
脱除蛋白的多糖溶液浓缩并去除有机试剂后,透析48 h,并间隔12 h更换一次蒸馏水,从而除去样液中的盐类及小分子物质,再浓缩、冻干,即可获得粗多糖。
1.3.2.3 阴离子交换柱层析
将经预处理的50 g DEAE-Sepharose Fast Flow用玻璃棒引流入层析柱(1.6 cm×30 cm),去离子水以12 mL/min流速冲柱5 min,再调流速至8 mL/min,平衡10 h以上。将10 mL 5 mg/mL粗多糖过0.45 μm水系一次性针头滤器。上样后,依次用去离子水与0.5~1.5 mol/L NaCl溶液梯度洗脱,控制流速2 mL/min,每管收集5 min,共收集80 管。同时采用苯酚-硫酸法[13]检测吸光度,并绘制洗脱曲线。收集样液,透析、浓缩、冻干,即可获得纯化多糖。
1.3.2.4 紫外吸收光谱测定
参照Kia等[14]方法并略加修改。取适量0.1%的纯化多糖溶液于比色皿中,测定样液在200~400 nm波长处的吸光度,以确定纯化多糖中蛋白质的脱除情况。
1.3.2.5 葡聚糖凝胶柱层析
参照董芳[15]的方法,采用进一步柱层析法检验纯化多糖是否为均一组分。将经预处理的适量Sephadex G-200填料灌入层析柱(1 cm×20 cm)并保证柱内无气泡或断层,以0.5 mL/min流速平衡。分别将5 mg/mL纯化多糖过膜,取1 mL上样,分别用去离子水和0.2 mol/L NaCl溶液洗脱,流速不变,每管收集3 mL,共收集40 管,依据溶液的吸光度绘制洗脱曲线。
1.3.3 小球藻多糖的结构性质
1.3.3.1 SEM观察
将适量已烘干的纯化多糖置于铜片上,粘贴固定好后放于镀金室内中,镀金操作完成后进行SEM并拍照,放大倍数为500、5 000。
1.3.3.2 HPLC分析
将2 mg纯化多糖与0.5 mL 2 mol/L三氟乙酸溶液混匀,120 ℃水解120 min,氮吹仪吹干。然后向5 μL 10 mg/mL单糖标准品与水解的多糖溶液中加入溶于甲醇的0.5 mol/L PMP试剂和0.3 mol/L NaOH溶液各0.5 mL,70 ℃水浴30 min,冷却至室温后,依次添加等体积0.3 mol/L HCl溶液与氯仿,振荡萃取,离心去除氯仿层,重复萃取3 次,水层过0.22 μm膜后,待上机。
色谱柱:Thermo ODS-2 C18柱(4.6 mm×250 mm,5 μm);流动相:0.1 mol/L pH 7.0磷酸盐缓冲液-乙腈(82∶18,V/V);流速1.0 mL/min;柱温25 ℃;进样量10 μL;波长245 nm。
1.3.3.3 刚果红实验测定
将纯化多糖配制成0.5 mg/mL溶液,依次取2 mL多糖、2 mL 60 μmol/L刚果红及不同体积的2 mol/L NaOH溶液,使得NaOH溶液终浓度分别为0、0.05、0.1、0.15、0.2、0.3、0.4、0.5 mol/L,静置15 min后测定不同浓度NaOH溶液中多糖最大吸收波长的迁移情况[16]。
1.3.4 小球藻多糖的流变学特性
为确定不同质量分数的纯化多糖(0.5%、1%、2%)在不同剪切速率(0.1~100 s-1)中的表观黏度。设置流变仪温度为25 ℃,测定并绘制纯化多糖的剪切速率与黏度的关系曲线[17]。
为确定不同质量分数的纯化多糖(0.5%、1%、2%)在不同角频率(0.1~100 rad/s)中的储能模量G’与损耗模量G”的变化。设置温度为25 ℃,剪切应变为0.1%,测定并绘制多糖的角频率与G’、G”的关系曲线[18]。
1.4 数据分析
应用Origin 8.6计算3 次重复实验数据平均值及标准偏差并绘制折线图。
2 结果与分析
2.1 分离纯化及鉴定分析
2.1.1 脱蛋白方法的选择
2.1.1.1 TCA法
由图1可得,蛋白质脱除率随TCA添加量的增加,先快速增加后基本维持于90%以上,最高可达95%。这是利用蛋白质阳离子在等电点与TCA结合形成不溶性盐[19]。但TCA添加量增加的过程中,多糖损失率在持续上升且当添加量在2%~5%时,上升趋势十分明显,可导致多糖损失率达30%以上。此结果也验证了之前报道的结论[20]。

图1 TCA法脱蛋白对小球藻多糖的影响Fig.1 Deproteinization efficiency of crude Chlorella polysaccharides with TCA
2.1.1.2 Sevag法

图2 Sevag法脱蛋白对小球藻多糖的影响Fig.2 Deproteinization efficiency of crude Chlorella polysaccharides by Sevag method
由图2可知,当采用Sevag法脱蛋白时,随着脱除蛋白次数的增加,蛋白质脱除率先缓慢增加后基本稳定于90%~95%之间。脱除蛋白次数小于6 次时,多糖损失率明显增加,随后基本保持于25%以下。目前已有研究[21]发现此法脱蛋白具有一定的优势,而且本实验结果也证明循环6 次Sevag法脱蛋白不但可以使得蛋白质脱除率达92.27 %,而且能够保证多糖损失率相对较低。因此,为了减少TCA试剂对多糖结构的降解,本实验拟采用Sevag法脱蛋白。
2.1.2 阴离子交换柱层析

图3 DEAE-Sepharose Fast Flow柱层析洗脱图Fig.3 Elution profile of Chlorella polysaccharides by DEAE-Sepharose Fast Flow column chromatography
由图3可知,小球藻粗多糖经DEAE-Sepharose Fast Flow柱层析可获得3 种不同级分的纯化多糖,分别命名为F1、F2、F3。其中F1为去离子水洗脱获得的多糖,冻干后为蓬松白色絮状物,而不同浓度(0.5、1、1.5 mol/L)NaCl溶液洗脱得到的F2与F3冻干后分别为淡黄色和乳白色蓬松絮状物。
2.1.3 紫外波长扫描

图4 纯化多糖的紫外光谱Fig.4 UV absorption spectra of the purified polysaccharides
溶液在200~300 nm范围内的吸收峰可以验证多糖中是否含大量的蛋白质及核酸[22]。从图4可以看出,多糖F1、F2、F3在260 nm与280 nm波长处几乎无明显的吸收峰,初步表明纯化多糖几乎不含蛋白质与核酸。
2.1.4 葡聚糖凝胶柱层析

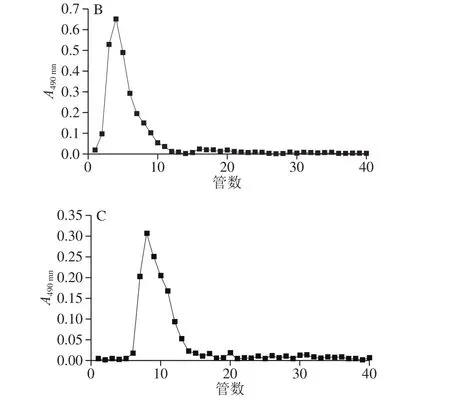
图5 Sephadex G-200柱层析洗脱图Fig.5 Elution profiles of the purified polysaccharides by Sephadex G-200 column chromatography
进一步葡聚糖凝胶渗透色谱上的对称单峰可以表明多糖接近于均质多糖[23]。本实验用Sephadex G-200层析柱分析。由图5可知,F1与F2的出峰时间接近,但与洗脱条件相同的F2相比,F3的出峰时间稍晚,由此推测F3的分子质量可能会略大于F2。从整体上看,纯化多糖均出现峰型基本对称的单峰,表明其接近于均一性组分。
2.2 结构性质
2.2.1 SEM分析

图6 纯化多糖的微观结构Fig.6 Microstructures of the purified polysaccharides
如图6A所示,F1表现为纤维细丝状,提高放大倍数可见明显的柱状结构,且表面平整,表明F1存在大量的分子或分子集团聚集形成的束。如图6B所示,F2呈现片状与碎屑状堆积,而放大5 000 倍时可见大量无规则交叉连接的立体网状结构,且表面粗糙,说明分子间的相互作用较强。如图6C所示,F3放大500 倍时呈现片状堆积,扩大放大倍数可见明显的片状结构表面含有不同层析的凸起,说明此多糖内可能存在一些具有一定排斥力的聚集体。
2.2.2 HPLC分析
10 种单糖标准品的出峰时间、回归方程及相关系数见表1,标准品、F1、F2与F3的单糖组成图谱分别见图7。不同纯化多糖的单糖组成及相对含量结果见表2,F1中相对含量较高的单糖分别为Gal(62.898%)、Glu(15.439%)和Man(12.665%),F2也含有较多的Gal(41.854%)、Fuc(13.741%)、Rha(10.199%),而F3主要是由Rha(32.33%)、Ara(39.55%)、Gal(10.82%)组成。纯化多糖F2与F3均不含Rib,与之不同的是F1不含GlcA。综上分析可知,小球藻多糖是一种以Gal为主的杂多糖,这与Song Hong等[24]所测结果相同,但不同于Qi等[25]的结论。后者认为Glu是优势单糖,并解释到微藻多糖的化学成分与小球藻物种和生长条件以及提取方法密切相关。

表1 单糖标准品的HPLC分析Table 1 Calibration equations and peak times of monosaccharide standards analyzed by HPLC


图7 单糖标准品与纯化多糖的HPLC分析Fig.7 HPLC profiles of monosaccharide standards and the purified polysaccharides

表2 纯化多糖的HPLC分析Table 2 Monosaccharide composition of the purified polysaccharides determined by HPLC
2.2.3 刚果红实验分析
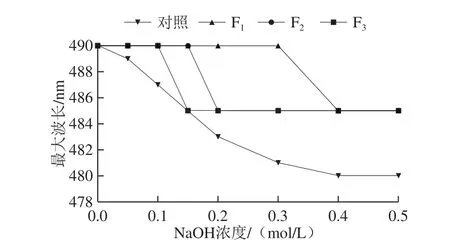
图8 纯化多糖最大吸收波长的变化Fig.8 Change in maximum absorption wavelength of the purified polysaccharides
在碱性介质中,刚果红染料可以与三螺旋结构的多糖反应形成络合物而导致溶液的最大吸收波长发生红移[26]。由图8可知,随着NaOH浓度的增加,纯化多糖的最大吸收波长发生了相同程度的蓝移,说明纯化多糖在水溶液中不呈现三螺旋结构,而表现为无规则线团链构象。
2.3 流变学性质
2.3.1 静态黏弹性分析
多糖为黏弹性材料,其流变行为除受内部微观结构的影响外,质量分数也是影响流变行为的重要因素之一[27]。由图9可知,3 种多糖的表观黏度都与质量分数和剪切速率有关,且呈现出质量分数的正向变化和剪切速率的负效应。其中F1的黏度变化范围相对较小,F2与F3的质量分数依赖性会随着剪切速率的增加而逐渐减弱。这是因为剪切力会破坏许多链间结构,导致系统黏度随剪切速率的增加而降低[28]。依据非牛顿流体的特征可以确定小球藻多糖溶液为非牛顿流体。

图9 纯化多糖的稳态剪切流动曲线Fig.9 Steady shear flow curves of the purified polysaccharides
2.3.2 动态黏弹性分析
由图10A可知,当角频率低于10 rad/s时,随着质量分数的增加F1的G’与G”也逐渐增加;当角频率高于10 rad/s时,质量分数增大虽然使得G”略有增加,但对G’影响不大。当角频率维持在0.1~10 rad/s之间,F1的G’与G”基本保持稳定,而角频率继续增加时,F1的G’与G”均呈现明显的上升趋势。
由图10B可知,当角频率不变时,F2的G’与G”与质量分数呈正相关。当F2的质量分数高于1%时,角频率对G’与G”的影响相对较小,仅在接近100 rad/s时轻微增长;当质量分数为0.5%时,随着角频率的增加,G’与G”展示出明显的大幅度增加趋势。
由图10C可知,与F2表现不同的是,当角频率在0.1~10 rad/s时,F3的G’与G”的质量分数依赖性不强,但质量分数高于1%时表现出质量分数依赖性增加;当角频率大于10 rad/s时,质量分数对G’与G”几乎不产生影响。当F3的质量分数不变时,F3的G’与G′随角频率的增加先稳定后显著增强。
在整个动态变化过程中,若G’>G”则表现为固体的弹性行为,而G’<G”表明溶液为类似液体的行为,出现交叉现象则说明多糖为凝胶型多糖[29-30]。综上所述,相同质量分数的纯化多糖始终保持G’>G”的状态,证明小球藻多糖为非凝胶型多糖。
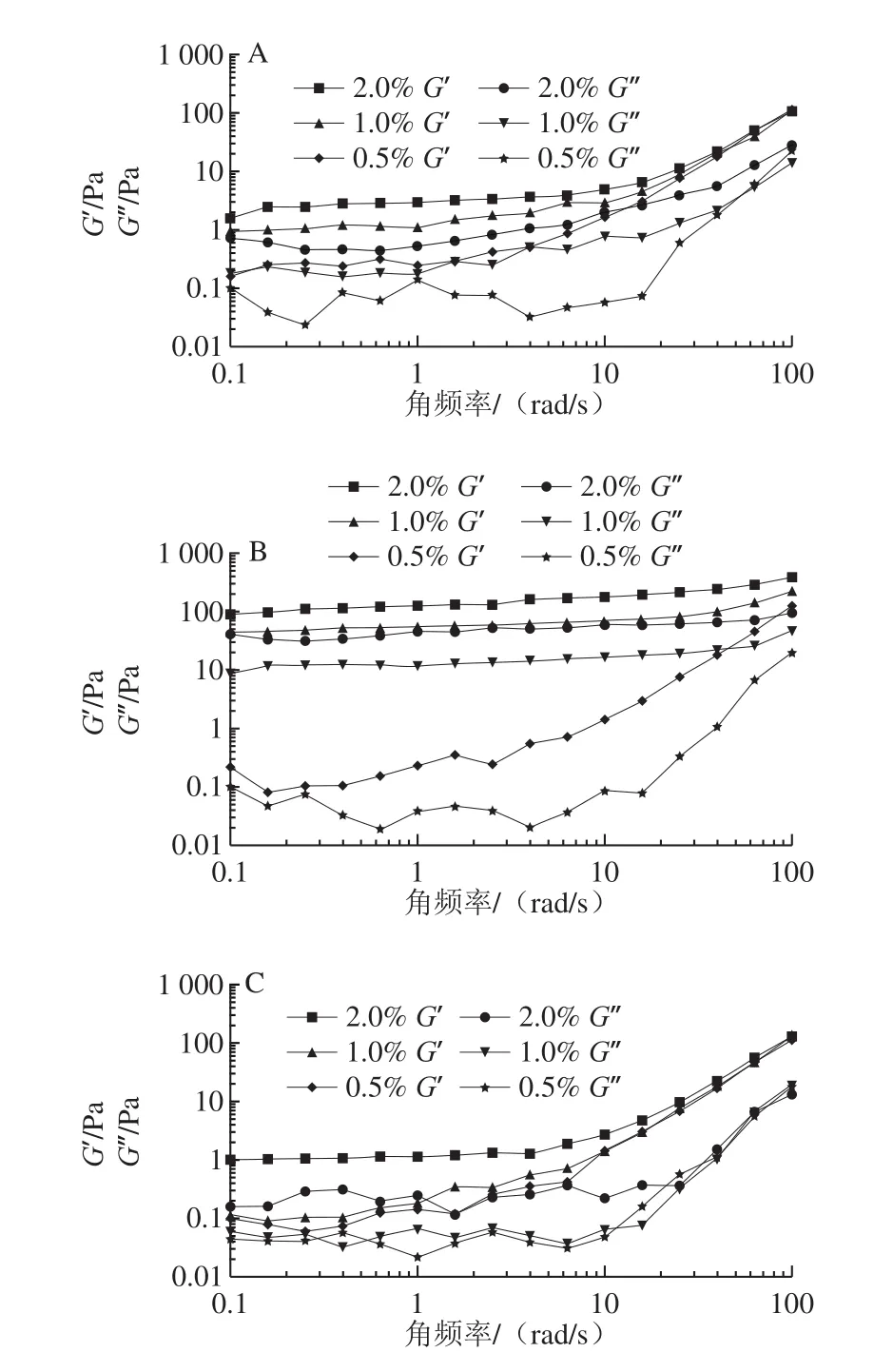
图10 纯化多糖的G’与G”的变化Fig.10 Change in G’ and G” of the purified polysaccharides
3 结 论
以蛋白质脱除率和多糖损失率为指标,通过对比分析TCA法与Sevag法对小球藻多糖的影响,发现循环6 次以上的Sevag法效果较好。紫外扫描与Sephadex G-200柱层析的鉴定确定脱除蛋白后的小球藻多糖经过DEAESepharose Fast Flow柱层析可以获得几乎不含蛋白质的3 种均一性多糖。SEM、HPLC以及刚果红实验结果显示,纯化多糖主要以片状或细丝状形式存在,在水溶液中不呈三维螺旋结构,且证实了小球藻多糖是以半乳糖为主的杂多糖。流变学性质分析探明小球藻纯化多糖为非牛顿流体,而且是一类非凝胶型多糖。
