利用全基因组重测序技术研究182份大麦和青稞的基因组结构变异
2020-10-28徐齐君王玉林杨春葆于明寨原红军
徐齐君,王玉林,杨春葆,扎 桑,于明寨,原红军
(1.省部共建青稞和牦牛种质资源与遗传改良国家重点实验室,拉萨 850002;2.西藏自治区农牧科学院 农业研究所,拉萨 850032)
青稞(Hordeumvulgarevar.nudum)作为主要粮食作物和牲畜饲料主要种植于青藏高原,青稞生产分别占西藏粮食和农区饲草总产的80%和60%以上,对保障西藏粮食安全、促进畜牧发展具有重要意义。其栽培历史可追溯到3 500年前。在过去的几年里,研究人员已经陆续公布青稞[1-3]和大麦(HordeumvulgareL.)[4]的细胞核和细胞器基因组,为基于参考基因组的结构变异分析提供了重要基础。结构变异(Structural Variation,SV)是一种基因组序列的重排现象,影响基因组片段大小超过50个碱基,不同于单核苷酸多态性(Single Nucleotide Polymorphisms,SNPs)和短插入/缺失(Insertions and Deletions,INDELs)。SV包括大片段的缺失(Deletion,DEL)、大片段的插入(Insertion,INS)、倒位(Inversion,INV)、易位和复制(Duplication,DUP)、拷贝数变异(Copy Number Variant,CNV),CNV为不平衡SV[5-6]。SV最初的检测方法是基于显微镜下的大规模染色体变化的核型观察方法,例如非整倍体[7-8]、染色体重排[9]和CNVs[10-11]。二代测序技术(Next Generation Sequencing,NGS)为SV的检测提供了一个前所未有的机会,可以从全基因组水平上阐明表型差异和复杂遗传变异的分子机制。与传统的实验技术相比,NGS技术已被广泛应用于动物和植物的遗传学研究,特别是SVs的研究。对人类的研究表明,SVs与精神分裂症[12]、癌症[13]和复杂的遗传紊乱疾病[14]有关。据估计,在人类基因组的遗传变异中,CNVs占比17.7%[15]。对植物基因组的广泛研究表明,SVs在植物适应进化和表型多样性中起着重要作用,如拟南芥[16]、黄瓜[17]、水稻[18-19]、高粱[20]和大豆[21]。基于NGS技术,正朝着遗传变异发现的新阶段前进,该阶段的重点是以单碱基分辨率来识别SVs在基因组中的分布式样。在过去的几年里,许多在种群水平上的重测序研究极大地丰富了人们对SVs机制和影响的认识。有研究表明,CNVs影响了大麦14个不同品种9.5%的编码基因[22]。西藏自治区农牧科学院农业研究所青稞遗传育种课题组之前的研究已对177份大麦材料进行了全基因组重测序,并进行了SNPs和INDELs分析,结果表明青稞起源于东方驯化大麦,最有可能在4 500年前至3 500年前,西藏青稞通过巴基斯坦北部-印度-尼泊尔传入西藏东南部;由于高原极端生态条件导致的奠基者效应,使传入后青稞遗传多样性快速降低,最终形成适应青藏高原生境的独特的青稞类群[2]。但之前的研究只对177份大麦和青稞的群体样本进行了SNP和INDELs分析,未对这些重测序数据进行基因组结构变异的分析。基于项目组之前已有的部分项目数据,结合从NCBI下载的5份数据,本研究分析182份大麦和青稞样品的重测序数据,平均测序深度约为12 X。基于高覆盖度基因组重测序数据,已经为大麦和青稞群体建立了高分辨率的基因组SV图谱,包括DEL、INS、INV和ITX(染色体内易位)。这些数据将为进一步的基因组学和遗传学研究,特别是探索相关基因的生物学功能起到重要作用。此外,作为一个有价值的补充,本试验产出的数据将促进并丰富大麦属物种的遗传变异研究。
1 材料与方法
1.1 试验材料
从存放在西藏自治区农牧科学院农业研究所的177份大麦/青稞样品中提取基因组DNA,这些样本来源于省部共建青稞和耗牛种质资源与遗传改良国家重点实验室青稞项目[2],数据已上传到NCBI数据库(BioProject accession number:PRJNA417220)。另外5份重测序数据从美国国立生物技术信息中心(NCBI)下载。本研究共获得182份大麦样品的数据,包括29份来自西藏(27份)、以色列(1份)和俄罗斯(1份)的半野生或野生大麦样品;从中国西藏、四川、青海、云南和浙江等省收集了70份地方品种的青稞;从日本(1份)、加拿大(3份)、澳大利亚(2份)、墨西哥(1份)、埃及(1份)、日本(1份)、叙利亚(2 份)和埃塞俄比亚(1份)共收集了12份地方品种的青稞;总共从中国西藏、青海、甘肃和四川收集了31份青稞育成品种,从加拿大收集了两份育成品种;其余34份大麦样品是在世界范围内收集的。
1.2 DNA提取和测序
使用快速植物基因组DNA分离试剂盒(Rapid Plant Genomic DNA Isolation Kit,中国上海,生工生物技术公司)从100 mg幼叶组织中提取基因组DNA,检测合格的DNA送往华大基因公司进行全基因组重测序。Illumina HiSeq 2000文库的构建按照制造商的说明书进行(Illumina,圣地亚哥,加利福尼亚州,美国)。使用Illumina cluster station进行合成,试验流程如下:模板杂交、等温扩增、线性化、封闭、变性和测序引物杂交。使用标准的Illumina base calling流程将荧光图像转换成序列。DNA文库插入片段为(454±39)bp,进行双端测序。Reads长度从 90 bp到150 bp不等。
1.3 基因组结构变异分析
Breakdancer[23]和SVDetect[24]是结构变异检测常用软件,专门针对双端测序数据进行开发,Breakdancer 和SVDetect可用于检测DEL、INS、INV、ITX(Intra-chromosomal translocation)和染色体间易位(Inter-chromosomal translocation),此外,SVDetect还可以用于检测DUP和CNV。Breakdancer运行速度较快,而SVDetect由于步骤较多,运行速度较慢。为保证结果的准确性,本研究使用Breakdancer和SVDetect对182份大麦和青稞群体中的主要SVs类型进行鉴定,筛选共有的SVs类型进行下一步分析。大麦的参考基因组序列从IBSC(http://webblast.ipk-gatersleben.de/barley_ibsc/downloads/150831_barley_pseudomolecules.fasta.gz;https://webblast.ipk-gatersleben.de/barley_ibsc/downloads/160517_Hv_IBSC_PGSB_r1_CDS_HighConf_REPR_annotation.fasta.gz)下载。此外,为消除未定位到染色体上的序列对SVs检测的影响,大麦参考基因组中chrUn的数据被排除在外。首先使用NGS QC Toolkit过滤原始数据并进行质控[25]。使用Bowtie 2软件采用默认参数,将质控过后的序列比对到大麦参考基因组[26]。SAMtools用于将SAM格式转换为BAM格式[27]。然后使用Picard包(https://broadinstitute.git hub.io/Picard/)去除重复序列,并使用Breakdancer[21]和SVDetect[22]软件执行SVs分析。Breakdancer采用默认参数。SVDetect采用以下参数:(1)ration filter参数为 0.35,coverage filter参数为6、0.05和2,depth filter参数为1.5和1.5,SVN filter参数为2,其它参数为默认值。本研究将上述2个软件的结果合并为最终的SVs结果。合并SVs条件如下:(1)多于4条支持序列或不少于3条不一致序列对;(2)SVs片段长度在100 bp和1 Mb之间[28];(3)重叠区长度大于等于25 bp并且2个SV位于同一染色体并属于同一变异类型,符合上述筛选条件的SVs被合并成一个SV[29]。本研究中只保留了Breakdancer和SVDetect软件检测到的主要SVs类型,包括DEL、INS、INV和ITX 4种SVs类型,并提取位于SVs变异区的基因用于后续功能分析。
1.4 基因功能分析
通过Gene Ontology Consortium(GO)所建立的数据库和参考基因组注释数据库(http://webblast.ipk-gatersleben.de/barley_ibsc/)对位于SVs变异区的基因进行功能注释和富集分析。
2 结果与分析
2.1 重测序数据和SVs在染色体上分布分析 结果
全基因重测序经数据质控后,总共产生 10 515 Gb数据。每个样本的重测序数据平均为 57.78 Gb,序列平均覆盖度为11.95X。SVs分析结果显示共获得了74 262个SVs,包括48 078个DEL(65%)、13 461个INSs(18%)、7 012个INV(9%)和5 711个ITXs(8%)(图1)。每个样本的平均SVs数量为408。在染色体分布式样中,7H的SVs的数量最多,占24.39%,其次是6H(14.07%)、2H(14.01%)、3H(13.07%)、4H(12.34%)、5H(11.50%)和1H(10.59%)。此外,与其他染色体相比,7H含有最高密度(28个/Mb)的SVs(图1)。

图1 SVs 在大麦染色体上的分布
长度分布显示,SVs片段长度主要分布在100 bp到1 kb之间,占所有SVs的64.26%。其中只有8.76%大于100 kb。大多数DELs(35 395/47 959)和INSs(11 466/13 180)长度从100 bp到1 kb不等。有趣的是,大多数INVs(3 139/7 012)和ITXs(2 252/5 708)的片段长度大于100 kb。与大麦参考基因组相比,在青稞的进化和选育过程中,很多基因组区域发生丢失,占所有SVs的64.93%(表1)。

表1 182份样本的SVs 长度分布
进一步为获得与SVs区域相关的可靠的基因集,从182份样本中选择了145份具有明确品种类型的样品,将其分成3组,包括野生大麦(30个)、青稞改良品种(33个)和青稞地方品种(82个)。对每份样品,采用以下条件下进行SVs区域相关基因的筛选:(1)基因区完全位于SV区;(2)每组80%以上样本具有相同的基因被保留。基于大麦参考基因的注释结果,分析结果显示18.72%的基因(7 440/39 734)位于SVs结构变异区。与大麦参考基因组相比,在野生大麦、地方品种和改良品种中共有380、357和391个基因在进化或选育过程中发生丢失。
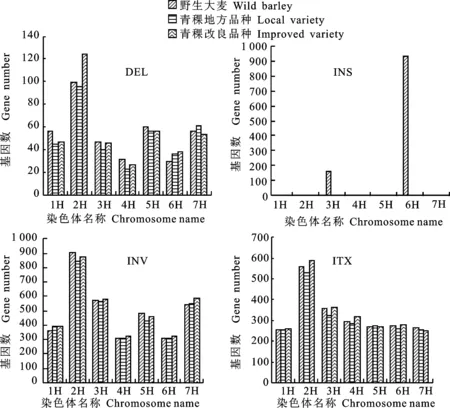
分析还发现大量基因插入到野生大麦群体中,占INSs的99.73%(1 093)。1个基因插入青稞地方品种中,2个基因插入到青稞育成品种中。INV和ITX相关基因的平均值分别为3 461和 2 257(图2)。进一步分析发现,基因的差异显示青稞育成品种、青稞地方品种和野生大麦中分别有62、23和37个独特的与DELs相关基因,280个与DELs相关基因在3个群体中共有(图2)。INSs分析结果表明,在青稞育成良品种、青稞地方品种和野生大麦群体中有2、1和1 093个独特的基因,在青稞地方品种中获得1个与INSs相关的基因。INVs结果表明,青稞育成品种、青稞地方品种和野生大麦分别有207、94和177个独有基因。与INVs相关基因数量最多。染色体分布结果显示,2H与DELs、INVs和ITX相关的基因数量最多。INSs主要分布在3H(158)和6H(936)染色体上,主要分布在野生大麦群体中,表明野生大麦含有较多的遗传资源,是遗传育种的重要基因资源库(图3)。
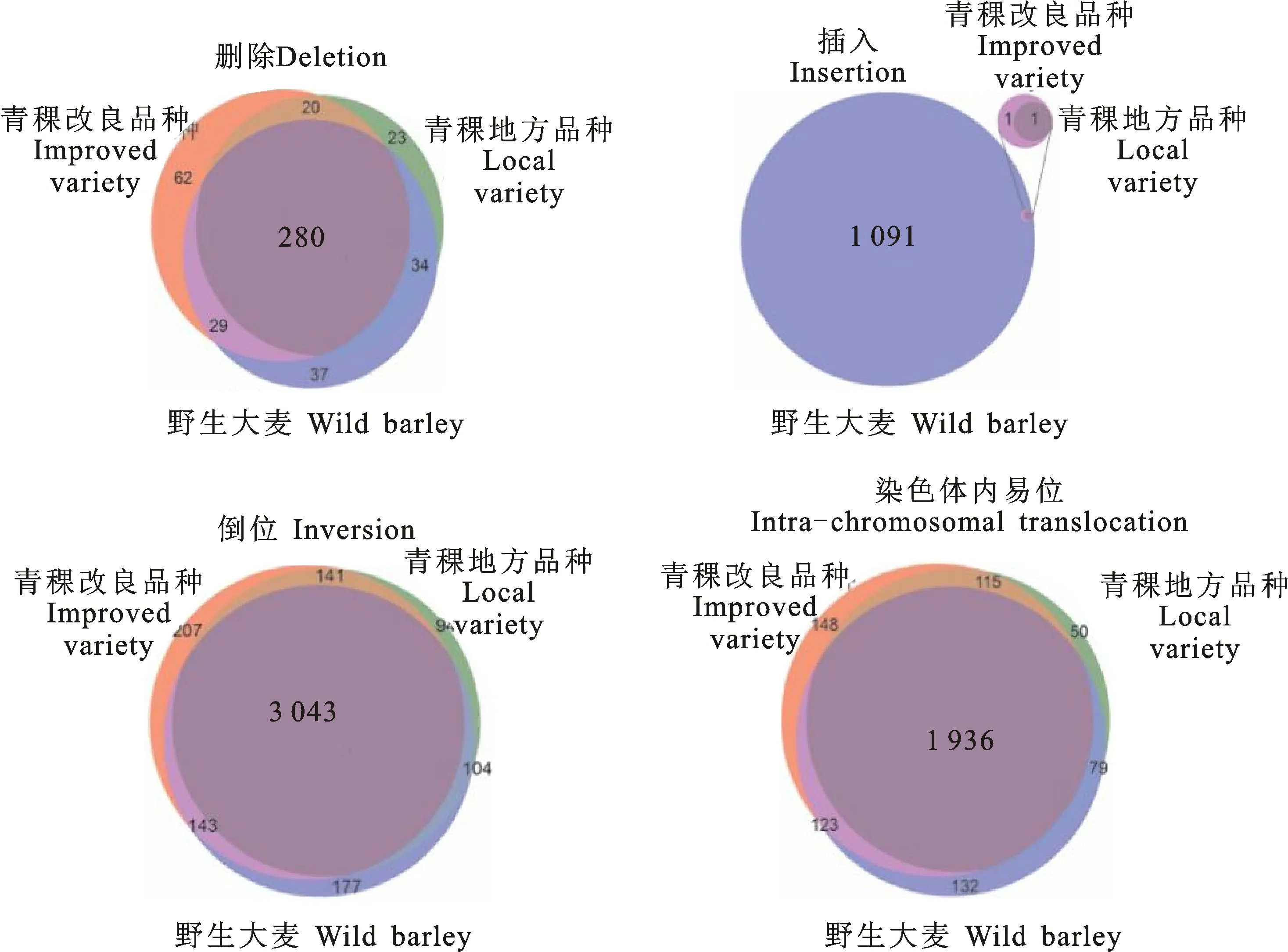
图2 与SVs相关基因的韦恩图
DEL.删除;INS.插入;INV.倒位;ITX.染色体内易位
2.2 位于SVs结构变异区基因的功能富集分析结果
与SVs相关基因的功能分析结果显示,筛选出的7 440个基因富集到945个GO term中,主要包括蛋白质结合(GO:0005515)、膜(GO:0016020)和氧化还原过程(GO:0055114)。GO功能富集最多的为INV(695),其次是ITX(583)、INS(331)和DEL(174)(图4)。此外,13个GO term与光系统相关,如光合作用(GO:0015979)、光周期开花调节(GO:2000028)和光合作用光反应(GO:0019684)。共有14个GO与植物应激反应过程有关。有趣的是,和大麦参考基因组相比,与真菌(GO:0050832)和细菌(GO:0042742)防御反应相关基因只在野生大麦中插入,与之相反,防御反应相关基因(GO:0006952)只在青稞育成品种发生丢失。在青稞育成品种中共有62个基因发生丢失,这些基因富集在48个GO term中,其中最丰富的是与蛋白质结合相关(44个基因)、锌离子结合相关(16个基因)和催化活性相关(16个基因)。大多数与DELs相关的基因属于大的多基因家族,包括糖基转移酶家族、WRKY、GRAS、锌指蛋白和五肽重复(PPR-like)家族。

图4 与SVs相关基因的GO terms 注释结果
3 讨 论
不断涌现的生物信息学工具和NGS测序技术的进步极大地促进了核苷酸变异的研究,尽管如此,对结构变异的研究在大多数物种中仍难以捉摸。本研究通过分析182份大麦和青稞群体全基因组重测序数据,用已公布的大麦基因组作为参考基因组,平均覆盖度为12 X。共获得74 262个结构变异,包含48 078个缺失(65%)、13 461个插入(18%)、7 012个倒位(9%)和5 711个染色体内易位(8%)。与SVs相关基因分析结果发现18.72%(7 440/39 734)的基因位于结构变异区域内。基因的功能分析发现,许多与结构变异相关的基因与光合作用和抗逆性相关,13个GO term与光系统相关,包括光合作用(GO:0015979)、光周期的开花调节(GO:2000028)和光合作用中的光反应(GO:0019684)。此外,还有14个GO term与植物应激反应过程有关。相关研究表明,与小尺度的核苷酸(<50 bp)变异相比,大片段的SVs在核苷酸变异中所占的比例更大,在人类基因组中,平均有8.9 Mb的碱基序列受到SVs的影响,而受到SNPs 影响的碱基序列只有3.6 Mb[30]。基于先前的研究结果,大麦基因组序列受到1.55 百万个SNPs变异的影响[2],其碱基序列长度远小于本研究中SVs 影响的碱基序列。结合与SVs相关基因的功能富集分析结果,说明SVs在青稞适应外界环境中重要性,亦表明物种基因组的多样性是其适应环境多样性所必需的遗传基础。
测序技术和生物信息学工具虽然在近几年取得了长足的进步,但核苷酸变异的研究目前主要集中在人类相关疾病的研究[30-31]。这种限制的主要原因是SVs是大规模的脱氧核糖核酸重组,带来了计算和生物信息学的挑战[32]。每种SV检测方法都有不同的优势和劣势,这取决于SVs的类型或SVs位点相关序列的性质[33]。本研究中,使用2种不同工具来分析SVs:Breakdancer[21]和SVDetect[22]。这些工具使用1种或2~3种基于参考基因组的比对方法,比如测序深度、双端比对或单端比对来检测SVs。这些方法本身很可能不足以揭示所有SVs[34]。本研究通过设置严格的筛选参数,合并以上2种软件的结果,以期望克服每种方法的弱点。
图2分析发现,INS相关基因与其余三种类型的分布模式不同,与INS相关的基因在野生大麦中大量聚集,这一结果与图3的结果一致,特别是在6号和3号染色体上。在野生大麦中发生插入结构变异,从另一个角度可以理解为在大麦参考基因组中发生大量丢失,我们推测在大麦参考基因组中,6号和3号染色体收到的选择压力远远强于其余5条染色体,这一现象也表明人工驯化或自然选择会引起基因组发生大片段的丢失,导致片段上基因资源的丢失,亦说明保护物种野生种质资源的重要性[35]。对SVs相关基因进行进一步分析发现,野生大麦、青稞育成品种和青稞地方品种之间存在如下差异,与DELs相关的基因中,青稞育成品种和青稞地方品种共有20个相同的基因,其中62个基因为青稞育成品种特有,23个基因为青稞地方品种特有,在INV和ITX结构变异类型中,青稞育成品种具有类似的规律,其特有基因数量都大于青稞地方品种和野生大麦,可能与青稞育成品种受到强烈的人工驯化作用相关,对玉米和水稻等驯化历史的研究显示,人工驯化会导致遗传多样性显著下降,即瓶颈效应[36-37],导致优异基因资源的丢失。育种工作者可以利用这些基因开展相关研究,特别是开展野生大麦中与SVs相关优异基因的研究,促进优良青稞品种的选育工作。
综上所述,本试验首次通过大规模全基因组重测序数据,对大麦基因组中的SVs进行了研究。由于分析工具和计算资源的局限,只选择了DEL、INS、INV和ITX 4种SVs类型,未能对大麦基因组中的CNVs进行分析,但本研究从各种类型结构变异中获得了新的生物学认识,并建立一个结构变异的综合数据集,为后续研究这些SVs相关基因对大麦和青稞生物多样性的影响提供支撑,可能有助于进一步理解大麦适应外界环境和非生物胁迫的分子机制。
