血浆替考拉宁浓度的反相高效液相色谱分析方法的建立Δ
2020-10-24杨云云王学彬高丽红张凌鹏张文静上海长海医院药学部上海00433
杨云云,蔡 璐,王学彬,高丽红,张凌鹏,张文静,王 卓(上海长海医院药学部,上海 00433)
替考拉宁为糖肽类抗菌药物,口服不吸收,一般采用静脉滴注或肌内注射给药。血清蛋白结合率为90%~95%,组织穿透性好,可进入白细胞内,不能进入红细胞、脑脊液和脂肪[1]。按照目前的指南推荐,替考拉宁和万古霉素是目前治疗革兰阳性菌感染的一线药物,特别是对于耐甲氧西林金黄色葡萄球菌(methicillin-resistantStaphylococcusaureus, MRSA)感染的效果较好[2-6]。由于替考拉宁的有效血药浓度与临床疗效密切相关,因此,合适的剂量有助于达到治疗相关血药浓度从而显现替考拉宁的疗效[7-8]。药动学/药效学(PK/PD)参数是抗菌药物临床合理应用的重要依据,目前替考拉宁的药品说明书不仅推荐了应用剂量,同时推荐了血药浓度,建议血药浓度不应<10 mg/L[9-12]。因此,建立测定替考拉宁血药浓度的方法对于提高临床合理用药水平十分重要。替考拉宁主要由5个结构类似的化合物TA2-1、TA2-2、TA2-3、TA2-4及TA2-5组成,其中TA2-2占总成分≥80%[13-14]。由于其组分复杂,建立快速、有效且准确的高效液相色谱法(high performance liquid chromatography,HPLC)相对困难,因此,主要通过测定TA2-2的浓度替代替考拉宁来建立快速有效的测定方法,现报告如下。
1 材料
1.1 仪器
LC-20A Prominence型高效液相色谱仪(日本岛津公司);C18色谱柱(美国Waters公司);XW-80 A型漩涡混合器(上海琪特分析仪器有限公司);移液枪(德国普兰德公司);UB-10型美国丹佛pH计(美国丹佛仪器公司);Eppendorf型离心机(上海琪特分析仪器有限公司);电子天平(10 mg~220 g)[赛多利斯科学仪器(北京)有限公司];SHB-ⅢA型循环水式多用真空泵(上海豫康科教仪器有限公司);DL-180A型超声波清洗器(上海之信仪器有限公司)。
1.2 药品与试剂
替考拉宁标准品100 mg(中国药品食品检定研究院);对羟基苯甲酸甲酯100 g(国药集团化学试剂有限公司);乙腈4 L(HPLC级)/瓶(TEDIA);磷酸二氢钠500 g(AR)/瓶(上海凌峰化学试剂有限公司);磷酸500 ml(AR)/瓶(上海凌峰化学试剂有限公司);人血浆(上海长海医院Ⅰ期临床实验室提供,血清置于-80 ℃条件下保存)。
2 方法与结果
2.1 色谱条件
色谱柱为Waters C18色谱柱(150 mm×4.6 mm,5 μm);保护预柱(ACE);流动相为0.02 mol/L的NaH2PO4·2H2O(H3PO4调pH至2.50)-乙腈(V∶V=74.5∶25.5);内标物为对羟基苯甲酸甲酯;流速为1.0 ml/min;检测波长为240 nm;进样量为0.020 ml;柱温为35 ℃。
2.2 溶液配制
2.2.1 替考拉宁标准品储备溶液:精密称取替考拉宁标准品20.00 mg,置于10 ml容量瓶中,加纯净水溶解稀释至刻度,摇匀,制成2 mg/ml的溶液。
2.2.2 内标溶液:精密称取对羟基苯甲酸甲酯5.0 mg,置于100 ml容量瓶中,加入乙腈溶液稀释后至刻度并充分混匀,制成0.05 mg/ml的溶液。
2.2.3 标准曲线配制:吸取标准品储备溶液50 μl,加入空白血浆950 μl,配制成质量浓度为100 μg/ml的样品A;吸取A溶液500 μl,加入空白血浆500 μl,配制成质量浓度为50 μg/ml的样品B;吸取B溶液500 μl,加入空白血浆500 μl,配制成质量浓度为25 μg/ml的样品C;吸取C溶液500 μl,加入空白血浆500 μl,配制成质量浓度为12.5 μg/ml的样品D;吸取D溶液500 μl,加入空白血浆500 μl,配制成质量浓度为6.25 μg/ml的样品E;吸取E溶液500 μl,加入空白血浆500 μl,配制成质量浓度为3.125 μg/ml的样品F[或称定量下限(lower limit of quantification,LLOQ)]。
2.2.4 质控样品配制:吸取标准品储备溶液50 μl,加入空白血浆950 μl,配制成质量浓度为100 μg/ml的样品A;吸取A溶液250 μl,加入空白血浆500 μl,配制成质量浓度为33.33 μg/ml的样品H;吸取H溶液250 μl,加入空白血浆500 μl,配制成质量浓度为11.11 μg/ml的样品M;吸取M溶液250 μl,加入空白血浆500 μl,配制成质量浓度为3.70 μg/ml的样品L。
2.3 血清样品的预处理
准确吸取空白血浆样本400 μl,置于离心管内,再向内加入50 μl内标储备液,加入乙腈600 μl,置于旋涡混合器混合1 min,放入离心机中以12 000 r/min的转速离心5 min。吸取100 μl上清液,置于样品瓶中,按“2.1”项下的色谱条件进样测定。
2.4 系统适用性试验
在空白血清中,加入2 mg/ml的替考拉宁储备液50 μl和0.05 mg/ml的内标溶液20 μl,涡旋混匀,按照血样处理方法制成血清样品测定液,按照“2.1”项下色谱条件进行测定。替考拉宁与内标物分离度良好,理论塔板数、重复性及拖尾因子均符合规定。替考拉宁的各组分保留时间合适,见图1。

图1 空白血浆中加入替考拉宁标准品和内标后的色谱图Fig 1 Chromatogram of blank plasma adding with teicoplanin standard and internal standard substance
2.5 线性范围评价
按照“2.2.3”项下方法制备标准曲线后进样,以校正因子f(TA2-2峰面积/内标峰面积)和X(TA2-2浓度)作线性回归分析,采用加权最小二乘法(X=1/C2),获得回归方程。替考拉宁在3.125~100 μg/ml范围内与校正因子f呈线性关系,回归方程为Y=0.016 175 6X-0.002 938 76(r=99.96%,n=6)。
2.6 准确度和精密度评价
按照“2.2.4”项下方法制备质控样品,取“2.2.3”项下LLOQ和高、中及低(样品H、M及L)4个浓度的质控样品各5份,每日检测1次,共检测3 d,结果见表1—2。相对误差(RE)=实测值/真实值×100%。批内和批间准确度均值一般应在质控样品标示值的±15%范围内;对于LLOQ,应在标示值的±20%范围内。
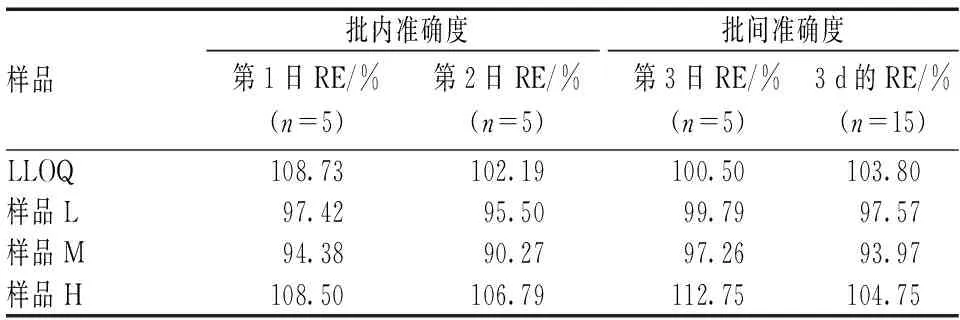
表1 批内及批间准确度Tab 1 Accuracy of intra-batch and inter-batch

表2 批内及批间相对标准差(RSD)Tab 2 Relative standard deviation (RSD) of intra-batch and inter-batch
2.7 提取回收率结果
制备3个浓度H、M和L含血浆的质控样品各5份,按照上述血浆处理方法处理,同时制备3个浓度H、M和L不含血浆的质控样品各5份,按照“2.1”项下方法进样,分别记录TA2-2和内标的峰面积。提取回收率计算公式:(质控样品峰面积/不含血浆质控样品峰面积)×100%。低、中、高和内标的提取回收率分别为102.86%±7.51%、100.52%±5.97%、97.65%±4.70%及100.92%±4.32%,能够满足测定的需求。
2.8 TA2-2及内标的稳定性
采用低、高浓度质控样品,平行3份,在预处理后及在所评价的条件储存后立即分析。由新鲜制备的校正标样获得标准曲线,根据标准曲线分析质控样品,将测得浓度与标示浓度进行比较,每个浓度的均值与标示浓度的偏差应在±15%范围内。将处理后的患者血清测定液置于-20 ℃保存,按照“2.1”项下高效液相色谱方法,于15 d取样进行含量测定,RSD为1.27%。表明-20 ℃条件下血清样品在15 d内是稳定的。
3 讨论
3.1 流动相及比例的调节
本研究流动相采用乙腈-磷酸二氢钠[15],通过不断调节流动相的比例,发现当乙腈所占比例增大时,出峰时间缩短,峰与峰之间的分离效果不佳;当磷酸盐所占比例增大时,也出现分离效果不佳的现象,并且有拖尾等问题出现。同时,将流动相pH范围设定在2.0~4.0进行实验,发现相对pH较高时,出峰提前;pH较低时,出峰延后,与文献报道相符[14]。通过不断调整pH、乙腈的比例,发现当NaH2PO4浓度为0.2 mol/L、pH为2.50时,乙腈与NaH2PO4的体积比在74.5∶25.5时,替考拉宁的各个峰都分离良好,保留时间长短合适,有利于实际临床样本的检测。
3.2 内标物的选择
本研究内标物的初始选择为哌拉西林钠,但是由于其稳定性差,在室温(25 ℃)条件下仅能维持6.5 h,-20 ℃条件下维持7 d,并且在后期的实验中出现双峰的现象,因此,后期研究的内标物调整为稳定性较好的对羟基苯甲酸甲酯溶,在室温条件下可维持2 d,-20 ℃条件下可维持30 d。由此可见,对羟基苯甲酸甲酯的稳定性明显优于哌拉西林钠,同时能够与目标峰分离。
3.3 柱温及流速的选择
流速增加,出峰时间缩短,但是重现性较差;流速降低,重现性尚可,但是出峰时间延长。1 ml/min为本研究的最佳流速。柱温对结果也有显著的影响,柱温升高,峰形会出现拖尾并且出峰时间延后。因此,在研究过程中需要保证流速和柱温的稳定。
综上所述,在本研究色谱条件下,替考拉宁能够在12 min内出峰,并且内标很稳定,与TA2-2分离度良好,能够作为临床的常规检测方法。
