骨髓衰竭疾病的体细胞突变在基因时代的临床意义
2020-10-14综述唐旭东审校
李 芮 综述 唐旭东 审校
1 骨髓衰竭的分子诊断
新一代测序(next-generation sequencing,NGS)已经对体细胞的大量基因和生殖系突变进行了筛选,这些基因的改变构成了髓样肿瘤如骨髓增生异常综合征(myelodysplastic syndrome,MDS)和典型的非克隆性骨髓衰竭综合征如再生障碍性贫血(aplastic anemia,AA)的主要致病原因。体细胞突变在临床管理中最明显的应用是可以更精确、更客观地对疾病实体进行分类或预测、并且在靶向药物开发和个性化治疗方面具有应用价值。与经典的X染色体灭活研究相比,体细胞突变可以作为克隆标记来定量克隆性造血。全外显子组测序的一个优点是可以全面分析突变的多样性和负荷,包括伴随突变和驱动突变。体细胞染色体畸变可以获得性基因组损害的突变总量,可以作为另一种度量形式。连续的NGS能够重建个体病例和动态的克隆结构(如微小残留疾病监测白血病的治疗效果)[1-5]。
2 MDS中体细胞突变和克隆结构
从理论上讲,MDS可能也存在如急性髓系白血病(acute myeloid leukemia,AML)的基因亚型。然而,体细胞突变谱的多样性和明显缺乏亚型特异性[6-8],导致MDS的体细胞突变谱分类非常困难,这要除外一些具有诊断性分子异常的体细胞突变如RUNX1、DDX41和CEBPA[9-11]和一些重现性分子异常如t(8;21) AML[12]。虽然存在以上问题,体细胞突变谱仍然在MDS中进行推广应用,并以形态学作为MDS诊断的金标准。早期事件也称为生殖系事件,根据此定义,它们既可以是克隆的(如在遗传性白血病中所见),也可以是非克隆的。MDS的一些基因突变是亚克隆的,而另一些基因突变可以是生殖系或亚克隆的。
初发AML中出现的特异性基因突变可能一直存在,从而导致疾病复发。最初的基因突变可能普遍存在,并不能早期预测疾病的发生,而某些次级次克隆事件却可能有预测疾病进展的作用。例如,在早期TET2突变后第2次发生的亚克隆TET2突变将导致骨髓增殖性疾病。同样,一些生殖系基因(如CEBPA、TP53和EVI1)可使剩余等位基因失活[13-16]。
一般来说,MDS可以通过克隆造血来启动,现在可以通过某些基因(包括DNMT3A、TET2、SF3B1等)的克隆突变来识别[17-18]。这种情况通常是无症状的,现在被称为潜质未定的克隆造血(clonal hematopoiesis of indeterminate potential, CHIP)。最终CHIP可能会产生不利影响。CHIP患病率随着年龄的增长而增加,尽管某些基因突变(如TET2)在老年人中的比例过高,而非典型DNMT3A突变构成了CHIP病变的主要部分。虽然所有的早期病变似乎都是由衰老因素导致的,但有些体细胞突变更容易引起无症状CHIP的疾病进展。对于某些体细胞突变来说,它们在克隆结构体系中的位置可能对预后的影响很大,而对于其他体细胞突变,它们在克隆结构体系中的位置并不那么重要。
迄今为止,可用的诊断和预后信息基于几项大型研究(表1)。已经发现临床相关的预后信息和各种体细胞突变的联系,如TP53、RAS基因家族、SETBP1和DNMT3A[6-8,19-21]。值得注意的是,个体突变可能具有不同的预后影响,特定突变的存在越来越多地决定了可能的治疗选择(如IDH1突变和AG-120;IDH2突变和AG-221;UTX突变和EZH2抑制剂;杂合TET2突变和5-氮杂胞苷/维生素C(可能);DNMT3A突变和S-腺苷甲硫氨酸(可能);JAK2 、CSF3R突变和鲁索利替尼;cKIT突变和阿西替尼和达沙替尼;Del 5q-和来那度胺;剪接体因子突变和H3B-8800)[22-24]。但在大多数情况下,单纯基因突变还不能确保疾病诊断的准确性。因此,与形态学金标准完全匹配的分子诊断模式尚未成功建立,当然未来的疾病分类方案可能更多地依赖于分子突变的划分而不是形态学的标准。
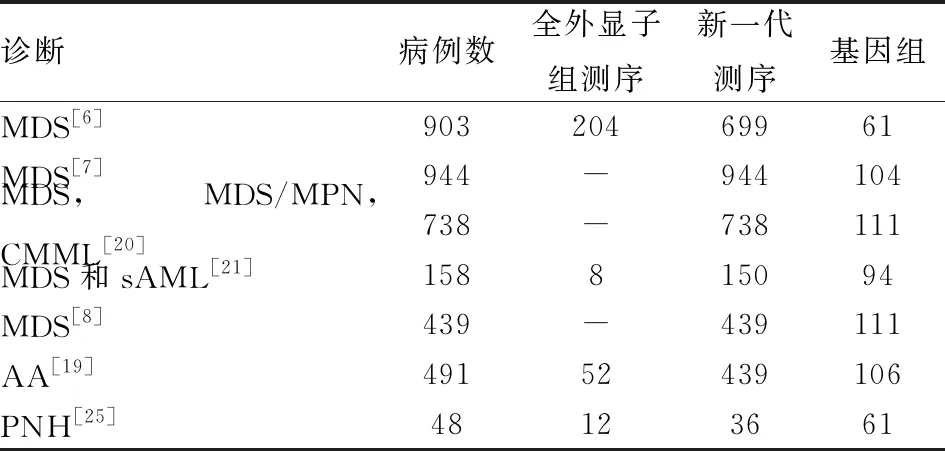
表1 骨髓衰竭的主要突变分析研究
3 AA和PNH中的体细胞突变和克隆结构
从理论上讲,正常的造血干细胞池(hematopoietic stem cell, HSC)的缩小可能导致寡核苷酸产生。例如,AA的克隆性染色体异常可以短暂出现,因为它们可以在恢复正常造血过程中通过正常HSC池的再扩张来缓解[25]。基于NGS的深度突变分析显示,以体细胞突变事件为特征的克隆可能会间歇性出现。相应克隆的变化可能取决于体细胞突变的类型[19]。初始呈现的某些克隆突变可以修复为正常克隆,或它们被选择和扩增,向相反方向发展,演进为MDS或PNH。通过流式细胞术鉴定PNH克隆,从而推断出AA克隆突变的存在[26]。
深度测序表明,PNH不是严格的单基因突变导致的。此外,一些PNH克隆显示出更复杂的亚克隆结构,这不仅涉及PIGA突变,还涉及典型MDS的其他骨髓基因突变。PNH的外在免疫选择理论并不能完全解释克隆演化的所有方面。例如,即使在免疫抑制剂治疗后,一些患者的克隆扩增仍在继续,而其他患者的PNH克隆保持稳定。已经观察到一些生殖系突变如PIGA突变和各种基因的亚克隆突变,类似于MDS突变[27]。在某些情况下,PIGA突变克隆与以嵌合体形式的其他突变克隆共存。
AA进展为MDS可以视作骨髓衰竭疾病的严重并发症,在临床上有着重要的治疗意义,10年内发生率为10%~20%[16,25-26]。大多数从AA演变而来的继发性MDS的特征是获得单体性7,通常是单个染色体畸变,提示预后不良[28]。如果骨穿无法获取足够的骨髓细胞数,单核苷酸多态性分析可能更有帮助[16]。在具有和不具有UPD6p的患者中发现了各种体细胞突变,如移码、无义和剪接位点突变,表明免疫选择(可能类似于PNH)可以导致HLA基因座中失活分子的克隆演进[16]。
克隆演进和分子学突变可能还基于其他潜在的机制。例如,一些具有-7/del(7q)的病例因为具有SAMD9和SAMD9L突变导致骨髓衰竭[29]。而在具有GATA2突变的患者中相对高频率的-7 / del(7q)表达,也可能涉及其他机制。临床上大多数进展为MDS的骨髓衰竭患者主要表现为难治或免疫抑制治疗反应不佳[28]。迄今为止,在AA转化为MDS的-7 / del(7q)保留等位基因上未发现复发性半合子,但在MDS中经常发现各种体细胞突变,包括SETBP1、CBL、RUNX1等[27]。对AA患者进行全外显子组测序或靶向深度测序,发现某些患者存在以体细胞突变为特征的克隆[19]。
这些突变除了PIGA中的突变外,还包括DNMT3A、CBL、SETBP1、TET2、ASXL1、BCOR和BCORL。某些体细胞突变频率的增加是否具有病理生理学意义还未可知。但总的来说,大多数体细胞突变仅存在于NGS评估的微小克隆中。在这些体细胞突变中的某几种突变虽不会持续存在但偶尔会扩增和消失,尤其是BCOR、BCORL和DNMT3A突变,这表明缩小的干细胞池具有募集选择的作用。此外,克隆突变的存在并不能预测MDS的进展;相反,只有特定的某些体细胞突变具有这种能力,而且随后的二次突变可以进一步加速MDS的演进过程。其他大多数体细胞突变则被免疫系统消除,或者通过与正常HSC竞争而消除。值得注意的是,在AA中发现的一些克隆可以表现为无症状的正常克隆,尤其在老年个体中常见。因此,这其实等同于AA患者中存在CHIP。
对AA的体细胞突变进行深度NGS追踪表明,亚克隆扩增已经存在于一定比例的AA患者中。而这些体细胞突变(如RUNX、SETBP1)可能引起后来的继发性MDS,并且表明del7不是初始缺陷,而是继发缺陷。AA发现克隆突变的一个重要科学意义是HSC存在动态演变以及对特发性AA起源理论进行了佐证。试想体细胞突变事件是如何引发肿瘤监视的免疫应答的:克隆突变可能与HSC相互作用并引起对HSCs的损伤,从而导致AA;机体的免疫系统消除了病理性克隆,并对最适宜体细胞突变克隆进行选择,如果这种被选中的克隆突变能够逃脱免疫监视,就会导致AA继发的MDS。
