大蒜转录组简单重复序列标记分析与分子标记开发
2020-10-14刘新雨
刘新雨,田 洁,2,*
(1.青海大学 农林科学院/青海省蔬菜遗传与生理重点实验室, 青海 西宁 810016; 2.青海大学 省部共建三江源生态与高原农牧业国家重点实验室,青海 西宁 810016)
大蒜(AlliumsativumL.)别名蒜头、胡蒜、葫,为百合科(Liliaceae)葱属(Allium)的栽培种,1~2年生草本植物[1-2]。大蒜栽培历史悠久且种植面积广,主要分布在山东、江苏、河南、广西、云南、四川等地区[3],在不同的生态环境下,所栽培的品种具有明显的区域性,形成了丰富的种质资源。丰富的种质资源是大蒜育种的基础,但因大蒜长期无性繁殖,受不同地区间引种和方言的影响,大蒜品种名和生产上用种混杂,导致大蒜品种更新慢、品种退化,使得大蒜栽培、育种工作难度增加,限制了大蒜产量和品质的提高[2,4]。因此,对大蒜资源进行评价与鉴定是首要工作。但是仅从形态学和解剖学等方面难以对所有的大蒜种质资源进行评价和有效鉴定[5-6]。
分子标记具有开发成本低、重复性好、信息量大等优点,被广泛应用于遗传图谱构建[7-8]、辅助育种[9-10]、物种亲缘关系鉴别和遗传多样性分析[11-12]等方面。目前应用于大蒜的分子标记主要有简单重复序列区间(inter simple sequence repeat, ISSR)标记、随机扩增多态性DNA(random amplified polymorphic dna, RAPD)标记、简单重复序列(simple sequence repeats, SSR)标记、扩增片段长度多态性(amplified fragment length polymorphism, AFLP)标记等[13]。陈昕等[5]利用RAPD和ISSR标记技术,对中国10个不同地区的大蒜品种进行了种质资源遗传多样性研究。周静[14]利用SRAP和SSR标记方法对40份中国大蒜品种进行了亲缘关系分析。孙亚丽等[15]利用已筛选出来的7对引物对55份大蒜资源进行UPGMA聚类分析认为,同一地域来源的种质大都被聚在一起。Ipek等[16]使用AFLP技术构建了大蒜低密度遗传图谱。韩曙等[17]利用RAPD标记对18个大蒜品种进行遗传多样性和亲缘关系分析,利用6个多态性明显的引物将18个品种分为2大类群。
SSR标记具有开发成本低、位点特异性高、通用性好等优点[18-19],已应用于花椰菜[20]、云锦杜鹃[21]、大麦[22]、大豆[23]、三角梅[24]等多种植物。相对而言,大蒜转录组SSR分子标记的开发较晚,Ma等[25]开发出8个多态性SSR引物,Cunha等[26]开发了10个新的SSR引物,陈书霞等[27]利用已开发的6对SSR引物对40份大蒜资源进行聚类分析和遗传多样性评价。目前基于转录组数据开发的SSR标记相对较少,限制了大蒜分子标记的研究,不利于大蒜分子标记辅助育种和遗传图谱的构建。本研究在大蒜转录组测序的基础上,对大蒜的SSR位点进行鉴别,并对其分布和组成特征等进行分析,以青海省农林科学院园艺所搜集到的35份大蒜种质资源为研究材料,对设计的部分SSR引物进行有效性与多态性鉴定,为后期种质资源评价、遗传多样性分析、遗传图谱的构建,以及分子标记辅助育种奠定基础。
1 材料与方法
1.1 数据来源
大蒜转录组数据来源于青海省农林科学院园艺所对乐都紫皮大蒜的高通量转录组深度测序。待大蒜幼苗苗高达10 cm后进行干旱胁迫处理,分别在处理0、3、6、9、12、15 d后进行取样,设置3次生物学重复,每个重复取长势一致的5株大蒜叶片做混样。取得后送至北京诺禾致源科技股份有限公司进行Illumina HiSeqTM测序,获得444 865条unigene,作为分析背景数据。
1.2 大蒜资源及其DNA提取
本试验以青海省农林科学院园艺所搜集的35份大蒜种质资源(表1)为基础进行SSR引物筛选、多态性鉴定与遗传多样性研究,其中独头蒜8份、多瓣蒜27份。每份样品取鳞茎100 mg左右,利用天根生化科技有限公司的DNAsecure新型植物基因组DNA提取试剂盒提取DNA,并检测所提DNA的浓度和纯度。DNA样品保存在-80 ℃备用。
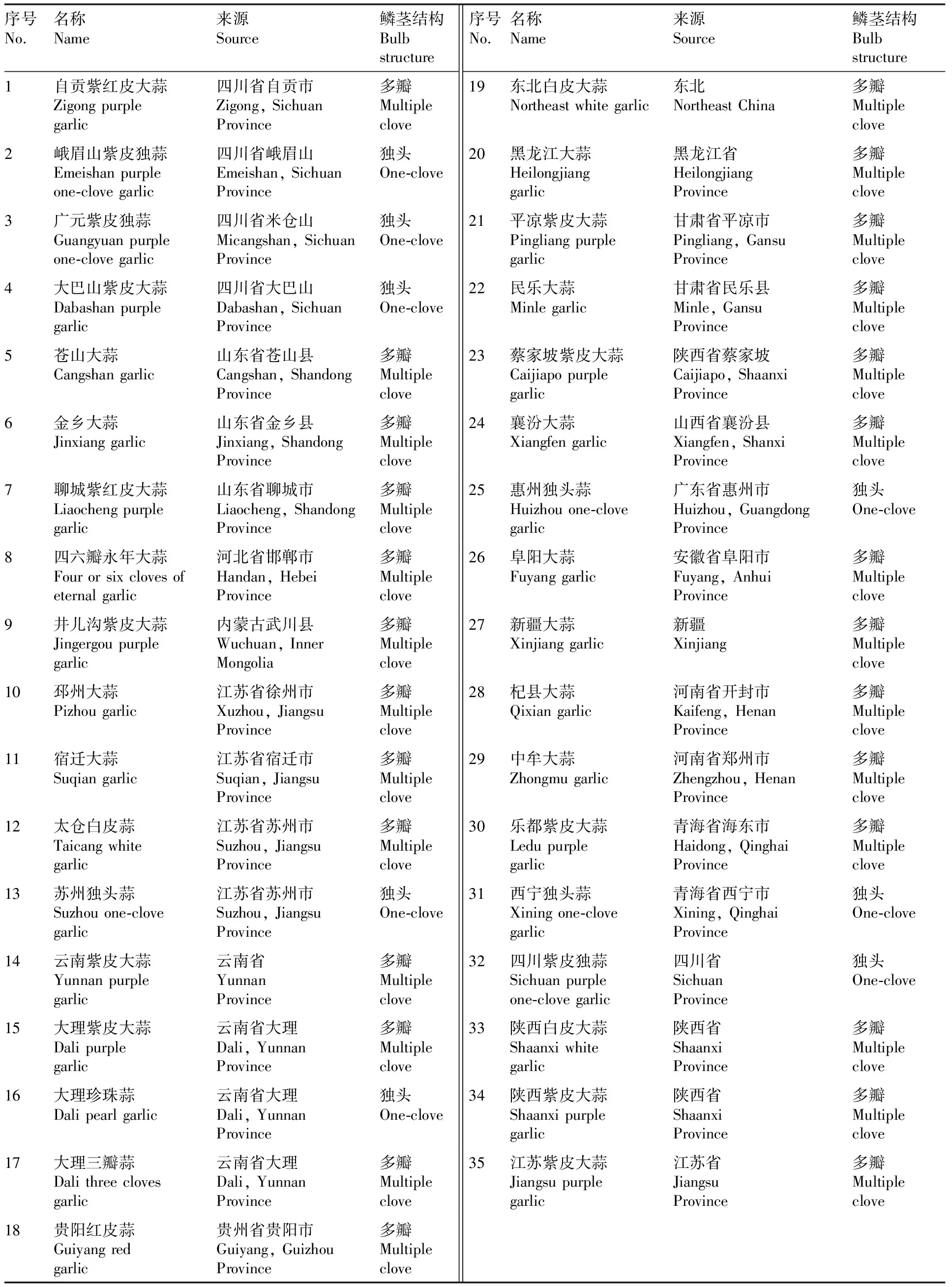
表1 三十五份大蒜种质资源信息Table 1 Information of 35 garlic germplasm resources
1.3 转录组SSR位点鉴别与SSR引物设计
采用软件MISA(1.0版,默认参数)对大蒜转录组序列的基因进行SSR检测,对应的各个unit size的最少重复次数分别为1-10、2-6、3-5、4-5、5-5、6-5(1-10表示以单核苷酸为重复单位时,其重复数至少为10才可被检测到;2-6表示以双核苷酸为重复单位时,其最少重复数为6。其他依此类推)。
用Primer 3.0进行SSR引物设计,并针对预测到的每一个SSR位点分别设计3组引物。引物设计的主要参数为:引物序列长度18~27 bp,预期扩增的片段长度120~300 bp,退火温度55~65 ℃。引物设计过程中避免出现引物二聚体、发卡结构与错配等引物二级结构。
为了验证引物的有效性和多态性,选择2个引物之间不发生互补,特别是在引物3′端,即使无法避免,其3′端互补碱基也应不大于2个碱基的14对SSR引物,送生工生物工程(上海)股份有限公司合成。
1.4 SSR引物筛选
利用PCR反应体系对选择的14对引物进行筛选。具体过程如下:首先,用3个大蒜品种的DNA混样筛选出条带清晰且重复性好的引物;然后,用35种大蒜资源对具有通用性的引物进行多态性验证。
PCR反应体系为10 μL:DNA模板1 μL,上下游引物各1 μL,2×TaqPCR Master Mix 4.5 μL,用ddH2O补充至10 μL。PCR扩增条件为:94 ℃预变性5 min;94 ℃变性30 s,54~58 ℃(因不同引物而异)退火30 s,72 ℃延伸30 s,30个循环;72 ℃延伸10 min。扩增产物4 ℃保存,用3%琼脂糖凝胶电泳检测,在90 V电压下电泳。
1.5 数据分析
采用人工读带的方法进行数据统计,稳定清晰可重复的条带记做1,相同位置不清楚或者无条带则记为0,构建矩阵,再用NTSYS 2.10软件采用UPGMA法绘制聚类图[28]。
SSR出现频率fc(%)=c/n×100,c为搜索到的SSR数量,n为无冗余转录本数量;SSR发生频率fC(%)=C/n×100,C为搜索到含SSR的无冗余转录本数量;SSR平均分布距离fN=N/c,N为无冗余转录本序列的总碱基数[29]。
2 结果与分析
2.1 大蒜转录组SSR位点的分布特征
大蒜转录组测序获得444 865条unigene基因序列,总长度486 687 980 bp,利用Misa软件对这些序列进行SSR位点鉴定。结果表明,全部unigenes中有102 421条序列包含SSR位点,发生频率(含SSR的基因数与总基因数之比)为23.02%,包含 1个以上SSR位点的序列有27 606条,共鉴定出141 132个SSR位点,出现频率(检出SSR数目与总基因数之比)为31.72%,其中16 593个复合SSR位点,平均每3.45 kb出现1个SSR位点。
由表2可知,在筛选出来的6种SSR基序中,以单核苷酸数量最多,为96 001个,占总SSR的68.02%;其次为二核苷酸,有34 051个,占总SSR的24.12%;三核苷酸、四核苷酸、五核苷酸、六核苷酸的数量相对较少,总共有11 080个,仅占总SSR的7.86%。不同类型的SSR位点出现频率与其比例相一致,以单核苷酸的出现频率最高,达21.58%;其次为二核苷酸,为7.65%;三、四、五、六核苷酸的出现频率相对较低。大蒜转录组中不同重复类型SSR位点的平均长度存在差异,单核苷酸重复平均长度为16.51 bp,二、三、四、五、六核苷酸类型SSR位点的平均长度分别为26.38、18.53、22.57、26.86、36.35 bp。
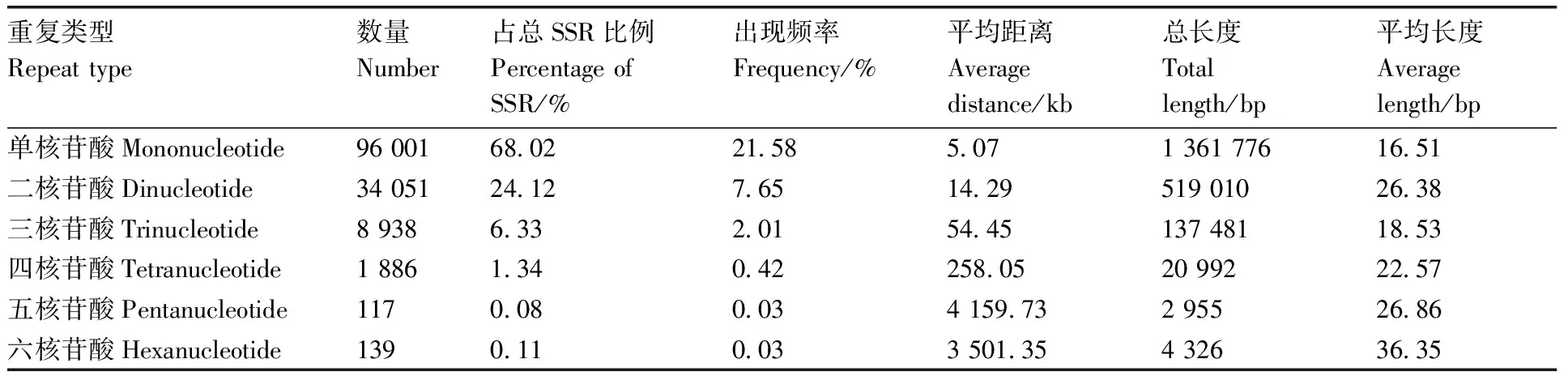
表2 大蒜转录组中SSR基序分布特征Table 2 Distribution characterisitics of SSR motifs in transcriptome of garlic
2.2 大蒜转录组中SSR基序类型与分布特征
由表3可知,大蒜转录组中SSR位点共有82种基序类型,以单核苷酸重复基序最多,有A/T和C/G两种类型,其中以A/T数量最多,有91 765个,占总SSR位点的65.02%;二核苷酸中以AT/AT数量最多,有17 460个,占总SSR位点的12.37%,其次分别为AC/GT、AG/CT、CG/CG,分别占总SSR位点的8.83%、2.63%、0.29%;三核苷酸中以AAT/ATT、AAG/CTT较多,分别有2 696个和2 386个,占总SSR位点的1.91%、1.69%;四核苷酸以ACAT/ATGT为主,有1 080个,占总SSR位点的0.77%;五核苷酸和六核苷酸重复基序类型所占比例均很少,其中五核苷酸重复类型共有15种,占总SSR位点的0.09%,以AAAAT/ATTTT为主;六核苷酸重复类型共有29种,占总SSR位点的0.1%,其中以AAGAGG/CCTCTT居多。
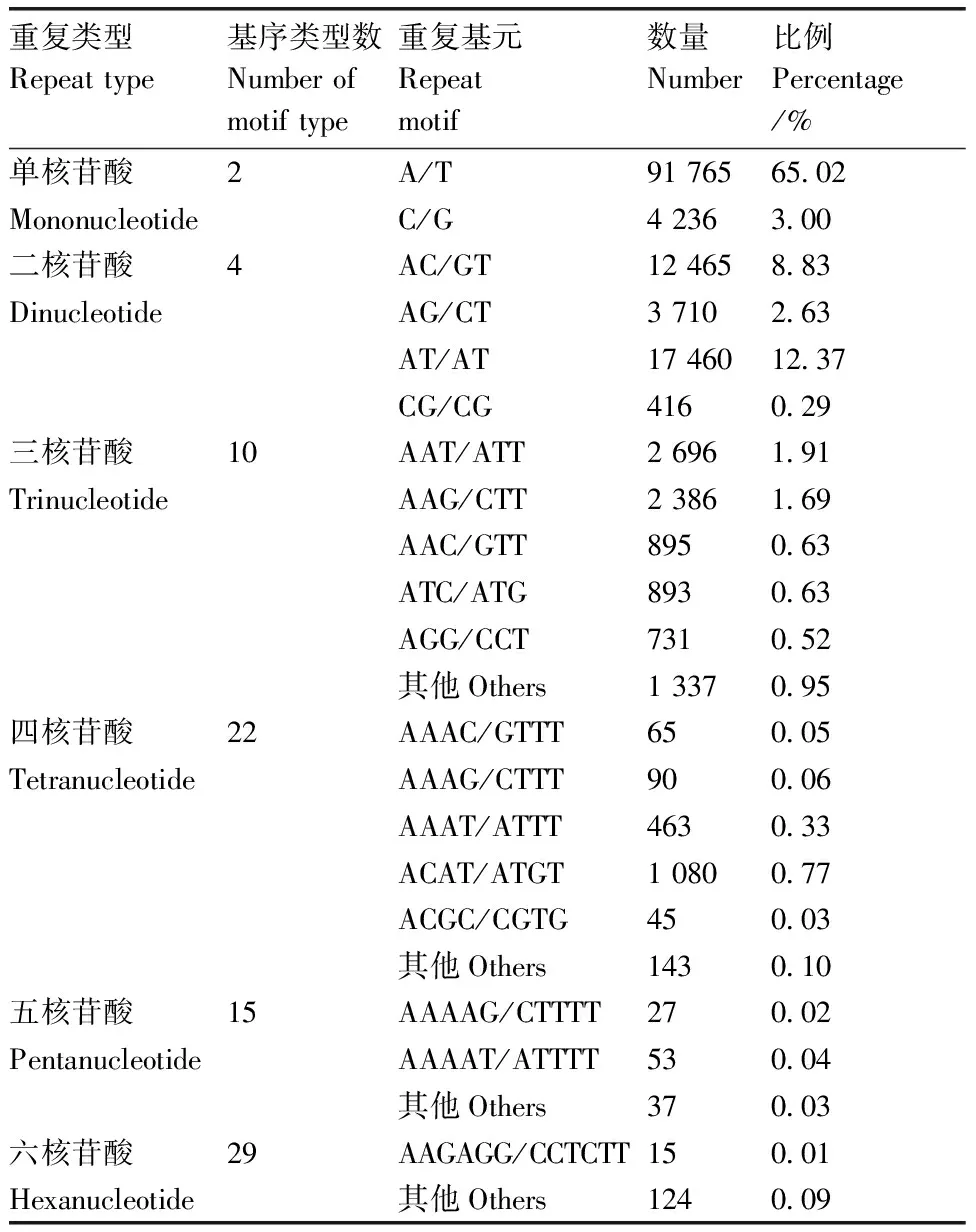
表3 大蒜转录组中SSR基序类型分布Table 3 Distribution of SSR motifs type in transcriptome of garlic
2.3 大蒜转录组中SSR重复次数与长度
大蒜转录组SSR基序重复次数随基序类型的不同而异。由图1可知,单核苷酸基序的重复次数主要集中在9~12次,其他核苷酸基序的重复次数主要集中在5~8次。总体来说,大蒜转录组SSR基序重复类型较多,其中重复5~30次的SSR位点均在1 000以上,以重复次数为10次的最多,共有30 407个,其次为11次,有15 399个。
已有研究表明,基序长度越长,其多态性越高[30],大于20 bp的重复序列有较高的多态性,重复序列长度在12~20 bp时表现出中等多态性,重复序列小于12 bp时表现为极低的多态性[19]。大蒜转录组SSR基序长度主要集中在10~325 bp,其中基序长度在10~20 bp的有80 915条,占总基序数的64.97%;基序长度在21~30 bp的有17 927条,占总基序数的14.39%;基序长度在31~40 bp的有8 345条,占总基序数的6.70%;基序长度在41~50 bp的有6 241条,占总基序数的5.01%;基序长度在51~60 bp的有4 089条,占总基序数的3.28%;基序长度大于60 bp的有7 022条,占总基序数的5.64%(表4)。由此可见,大蒜转录组SSR位点基序主要集中在10~20 bp,属于中度多态性。

数字5~30代表核苷酸基序的重复次数。The numbers 5-30 referred to repeat counts of nucleotide motifs.图1 大蒜转录组中SSR不同基序重复次数分布Fig.1 Distribution of various SSR motifs with different numbers in transcriptome of garlic

表4 大蒜转录组SSR位点重复序列的长度分布Table 4 Distribution of length of repeat sequences of SSR loci in transcriptome of garlic
2.4 大蒜SSR引物多态性分析
根据大蒜转录组SSR位点,本研究共设计出125 616对引物,为了验证其有效性,随机选择了14对引物对金乡大蒜、大理珍珠蒜、大理三瓣蒜3个大蒜品种的DNA混样进行PCR扩增,其中12对引物能够扩增出清晰的条带,引物有效率为85.71%。利用35份大蒜资源对筛选出的12对有效引物进行多态性鉴定,结果表明,其中6对引物具有多态性差异(表5),占有效引物的50%;每对引物的扩增条带为1~9条,共37条;扩增得到的多态性条带共26条,每对多态性引物平均产生4.33条条带;其中引物11多态性为66.67%。图2所示是引物11在35份大蒜资源中的扩增情况。

表5 大蒜6对SSR多态性引物信息Table 5 Information of 6 pairs of pleomorphic primers developed from garlic
2.5 聚类分析
用6对具有多态性差异的引物对35份大蒜资源进行聚类分析,结果表明,35份大蒜资源遗传相似系数介于0.67~0.97,在遗传相似系数0.756 9处,35份大蒜资源可被分成5大类群(图3)。聚类结果表明,大部分来自同一地理区域或相近地理区域的品种聚在一起,比如第Ⅰ类的8份资源主要来源于西南地区,第Ⅱ类的7份资源主要来自西北地区;但也有少部分地理距离相距较远的被聚在一起,如来自安徽的阜阳大蒜(26号)与来自东北地区的资源均被聚到了第Ⅴ类,这可能是由于地区间的引种所造成的。UPGMA聚类结果表明,大蒜区域间存在着基因交流,并且SSR标记能够有效地对不同地区来源的大蒜资源进行区分,并进行亲缘关系的鉴定。

M,DNA marker;1~35,表1 中35份大蒜资源的序号。下同。M, DNA marker; 1-35, Number of 35 garlic germplasm resources in Table 1. The same as below.图2 引物11在35份大蒜种质资源中的多态性Fig.2 Polymorphisms of primer 11 in 35 garlic germplasm resources
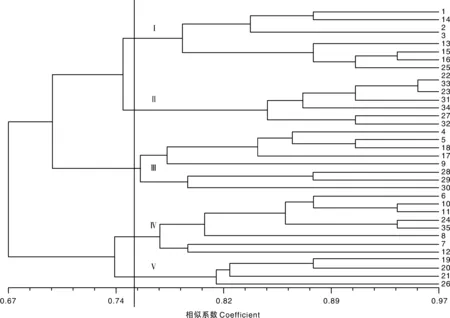
图3 三十五份大蒜种质资源的聚类分析Fig.3 Clustering analysis of 35 garlic germplasm resources
3 结论与讨论
近年来,随着高通量测序技术的快速发展,转录组测序成本大大降低。SSR分子标记以其成本低、通用性好等优点已应用于多种植物。大蒜的栽培面积广,种质资源丰富,但由于大蒜地区间品种交流频繁使大蒜种质来源混杂,这就极大地增加了大蒜种质资源分类和育种的难度。相对于形态标记,分子标记可以不受环境的影响,且SSR分子标记对大蒜基因组检测多态性水平高,能够有效进行遗传多样性分析和亲缘鉴定等,从而提高大蒜育种的效率。本研究以大蒜转录组测序获得的444 865条unigenes为基础,其中有102 421条序列包含SSR位点,SSR位点发生频率为23.02%,较韭菜(20.72%)[29]、款冬(20.30%)[31]、山茶(19.52%)[32]、棉花(3.05%)[33]的发生频率高,但低于牡丹(29.19%)[34]、莲雾(38.36%)[35]、蓝靛果忍冬(32.51%)[36]。不同研究中SSR位点发生频率不同,可能是转录组测序方法、SSR位点筛选条件、开发软件和物种间差异等造成的。
本研究中大蒜转录组SSR位点的重复类型存在着严重的偏倚现象。以单核苷酸、二核苷酸和三核苷酸重复基序为主,占总SSR总数的98.47%,四、五和六核苷酸重复基序所占比例很小,仅为1.53%。这与贺丹等[34]的研究结果相一致。已有研究表明,三核苷酸是大多数植物的主导重复基序[28,37],但本研究中以单核苷酸(68.02%)重复基序最多,其次是二核苷酸(24.12%)和三核苷酸(6.33%)重复,这与前人在莲雾[35]、连翘[38]中的研究相一致,这可能与转录组unigenes中包含更多的非编码区(UTR)信息有关[35],或者是不同植物基因组在进化过程中经历的时间不同[34]。单核苷酸重复基序中以A/T数量最多,这与前人对莲雾[35]、芙蓉李[39]等的研究得出的结论相一致。二核苷酸中以AT/AT数量最多,这与前人研究的二核苷酸主要重复类型为AG/CT不一致[35,28,40],这可能与乐都紫皮大蒜栽培品种叶的SSR特性对大蒜转录组中SSR位点的影响有关。
本研究选择的14对引物中有12对引物能进行有效扩增,其中有6对具有良好的多态性,多态率达50%,平均每对多态性引物可扩增得到4.33条条带,表明本研究开发的SSR引物扩增效率较高,具有较高的可用性;6对引物的多态率高于油梨的10.79%[41],低于花椰菜的60.71%[20],说明不同品种间SSR分子标记结果存在着差异。本研究利用6对多态性引物将35份大蒜资源分成了5大类群,且来自同一地区的品种大多被聚集到同一类群中,表明本研究开发的SSR引物在一定程度上能够有效地区分不同来源的大蒜资源,具有较高的应用价值;但不同地区来源的资源也可能被聚集到一起,如来自江苏的苏州独头蒜(13号)与来自云南的云南紫皮大蒜(14号)等均被聚集到第Ⅰ类群,这可能是由于大蒜区域的相互引种所造成的,这也说明了大蒜种质来源复杂;大理三瓣蒜(17号)与大理紫皮大蒜(15号)和大理珍珠蒜(16号)虽然均来自云南且地理位置较近,但却被分到2大类群中,表明大蒜本身具有复杂的遗传多样性。本研究发现大蒜SSR标记基本可以用于亲缘关系鉴定和判断品种间是否存在基因交流等。
