生物转化脂肪酸合成ω-羟基酸和ω-氨基酸研究进展
2020-09-29丁良怡种刚刚潘江许建和
丁良怡,种刚刚,潘江,许建和
(华东理工大学生物反应器工程国家重点实验室,上海200237)
引 言
脂肪酸是一类由碳、氢、氧组成的,具有较长烃链的羧酸类化合物。脂肪酸是自然界最为丰富的物质之一,而富含脂肪酸的油脂成为化工行业最重要的可再生原料[1]。人类对不可再生化石资源的过度依赖,促进了全球生物柴油产业的发展,植物油的综合利用问题也日益突出[2]。植物油广泛存在于自然界中,生产量巨大,然而目前大部分仍被简单用于生产食用油和饲料添加剂,只有一小部分用于制造生物柴油和生物基化学品[3]。大多数植物油由1 mol 甘油和3 mol 脂肪酸组成,后者主要包括不饱和脂肪酸油酸和亚油酸,其中含有两类活泼的官能团,即末端羧基以及若干碳碳双键,对这些基团的各种修饰反应是其工业用途的基础。工业上,目前主要对脂肪酸的羧基进行反应,转化为脂肪醇类、酯类、酰胺类或胺类等,进一步用于生产表面活性剂、增塑剂、涂料等产品[4]。随着生物化工技术的发展,利用具有良好选择性以及反应条件温和的生物催化工艺实现脂肪酸的功能化衍生和精细化增值正受到越来越多的关注。
对脂肪酸进行一系列氧化降解、加氢还原以及环氧化、胺化等反应可以生成很多种脂肪酸衍生物,可用于生产各种各样的化学品。目前大多数油脂类化学品的生产采用了高温高压、强酸或重金属催化剂、有毒催化剂(如臭氧)等苛刻的反应条件,且步骤烦琐、选择性较差[5],所以人们越来越倾向于寻求一种更绿色和可持续的新工艺,以便采用更温和的反应条件,减少安全隐患以及减少工业废物,降低对环境的影响[6]。因此,亟需开发新的反应路径为脂肪酸的衍生化提供新的思路,以提升脂肪酸的应用价值。
如图1所示,采用生物酶催化法,以植物油中丰富的油酸、亚油酸或蓖麻油酸等长链不饱和脂肪酸为底物,通过水合酶、醇脱氢酶、单加氧酶、脂氧合酶、酯酶、脂氢过氧化物裂解酶、胺脱氢酶以及转氨酶等多酶协同催化,可生成不同链长的二醇、二酸、二胺以及ω−氨基脂肪酸(ω−AmFAs)、ω−羟基脂肪酸(ω−HFAs)等脂肪酸衍生物,它们可以作为单体合成聚酰胺、聚酯等系列聚合物[7−8],用于生产塑料、润滑油、香料、涂料、燃料等产品,此外它们在一些药物生产中也是重要的合成前体[9]。本文主要概述了近年来植物油以及长链脂肪酸转化为中等链长(C6~C12)ω−HFAs和ω−AmFAs的各种生物催化路线,重点介绍了目前ω−HFAs 和ω−AmFAs 的酶法合成研究进展。
1 ω−羟基脂肪酸
羟基脂肪酸(HFAs)含有羧基以及一个或多个羟基,碳链可以是饱和或不饱和的[10]。与普通脂肪酸相比,HFAs 因具有更高的黏度和反应活性等特殊性质而备受关注[11]。根据羟基所处的位置,HFAs 也可以简单分为α−HFAs,β−HFAs,ω−HFAs 以及中位HFAs。其中,ω−HFAs 由于其羟基连接烃链最末端的碳原子,从而为合成高分子材料提供了最长的碳链骨架,可用于生产优质的绿色合成纤维[10]。近年来,为扩大植物油的高值化利用范围,植物油中富含的脂肪酸如油酸、亚油酸、蓖麻油酸等被广泛研究和开发,主要是应用生物催化技术合成工业所需的ω−HFAs。
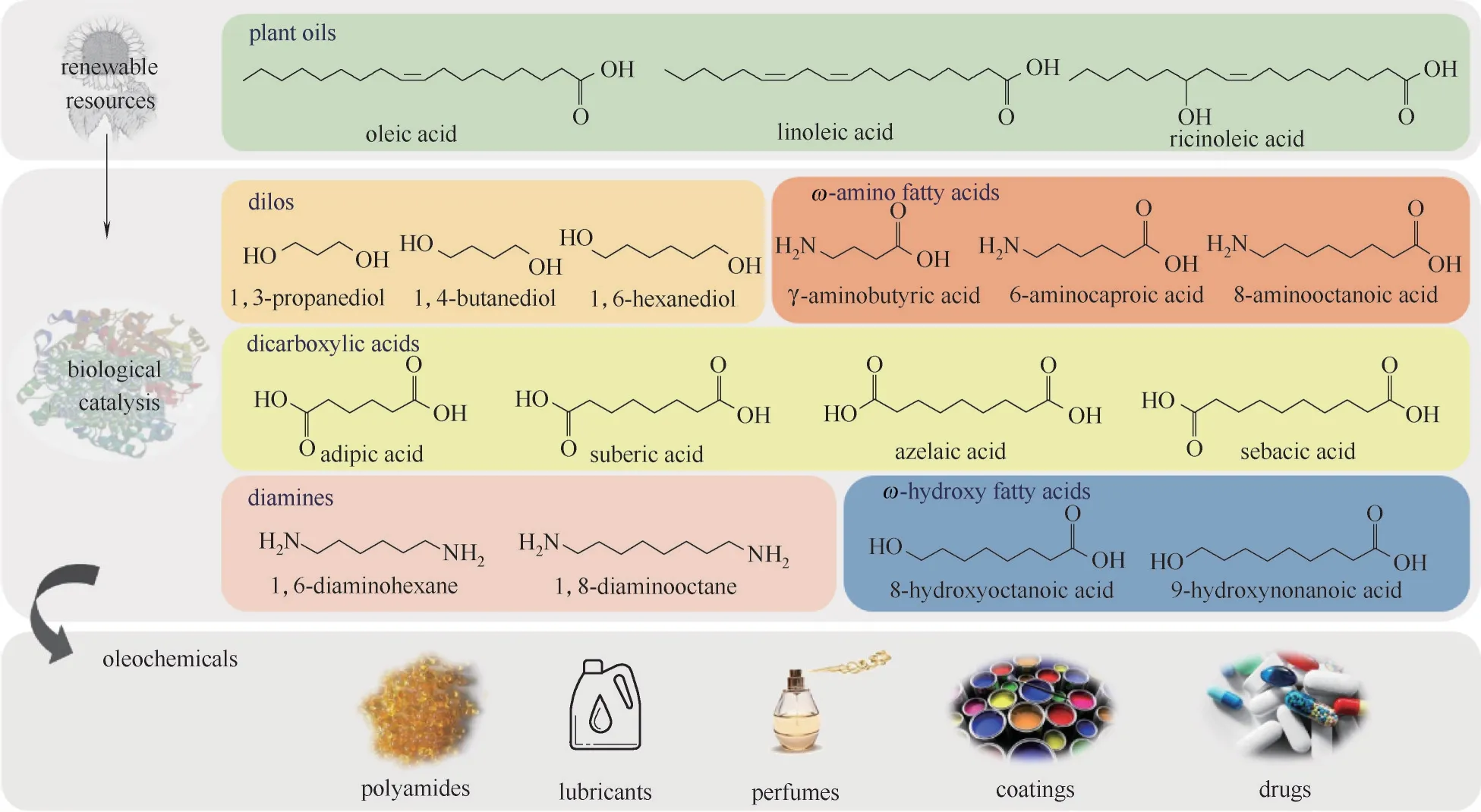
图1 酶催化脂肪酸转化生成多种功能化合物及其应用价值Fig.1 Enzyme−catalyzed conversion of fatty acids into various functional compounds with diverse application values
ω−HFAs 的合成可以分为两类:脂肪酸的末端C—H 氧化以及脂肪酸碳链内部的氧化裂解[3,12]。其中,碳链末端的C—H 氧化反应主要由P450 单加氧酶或烷烃氧化酶AlkBGT 催化;而碳链内部的氧化裂解反应主要由拜耳−维利格单加氧酶(Baeyer−Villiger monooxygenases,BVMOs)或脂氧合酶及裂解酶催化,这也是长链不饱和脂肪酸转化为中链ω−HFAs 中的一个常用反应。本节主要介绍植物油转化为ω−HFAs的多酶级联反应,汇总了油酸、亚油酸和蓖麻油酸这三大类植物油通过上述第二类反应转化为饱和、不饱和ω−HFAs 的生物催化级联反应(表1)。
1.1 饱和ω-羟基脂肪酸的合成
脂氧合酶可以在亚油酸、亚麻酸等脂肪酸的双键处加氧生成过氧化物,再通过裂解酶断裂生成末端醛酸。以亚油酸为例,为获得中间产物9−醛基壬酸以进一步合成9−羟基壬酸及其衍生物,Otte 等[13]运用来自Solanum tuberosum 的9S−脂氧合酶(9S−lipoxygenase)StLOX1 以及来自Cucumis melo 的9/13−裂解酶(9/13−hydroperoxide lyase)Cm9/13HPL,将亚油酸转化为9−醛基壬酸(图2)。由于9/13HPL 具有严格的S−构型选择性,所以需要采用高S−选择性的LOX 进行催化。该研究发现,随着底物上载量的提高和反应时间的延长,可能导致中间产物的异构化,使得产物得率明显降低。
脂氧合酶催化亚油酸不仅可以催化C9位也可以C13位加氧生成过氧化氢衍生物,然而其作为植物酶在微生物细胞中表达量低、活性差,影响了其在重组微生物细胞中的生物转化效率,因此筛选高活力的LOX 具有重要的研究意义。2020 年,本课题组[20]报道了三个新的来源于蓝藻的脂氧合酶,分别是CaLOX (来源于Calothrix sp. HK−06)、RiLOX (来源于 Rivularia sp. PCC 7116) 和TbLOX ( 来 源 于Tolypothrix bouteillei VB521301),其中CaLOX 和RiLOX是迄今报道中活性最高的LOXs,纯酶的比活力分别达到73.1 和68.8 U·mg−1。RiLOX 在大肠杆菌中异源表达时,体系酶活可以达到38.3 U·ml−1,它催化亚油酸生成13−过氧氢−9,11−(Z,E)−十八碳二烯酸(13−HPOD),后续使用NaBH4还原可得到13(S)−羟基−9,11−(Z,E)−十八碳二烯酸(13−HODE),时空产率高达1056 g·L−1·d−1。
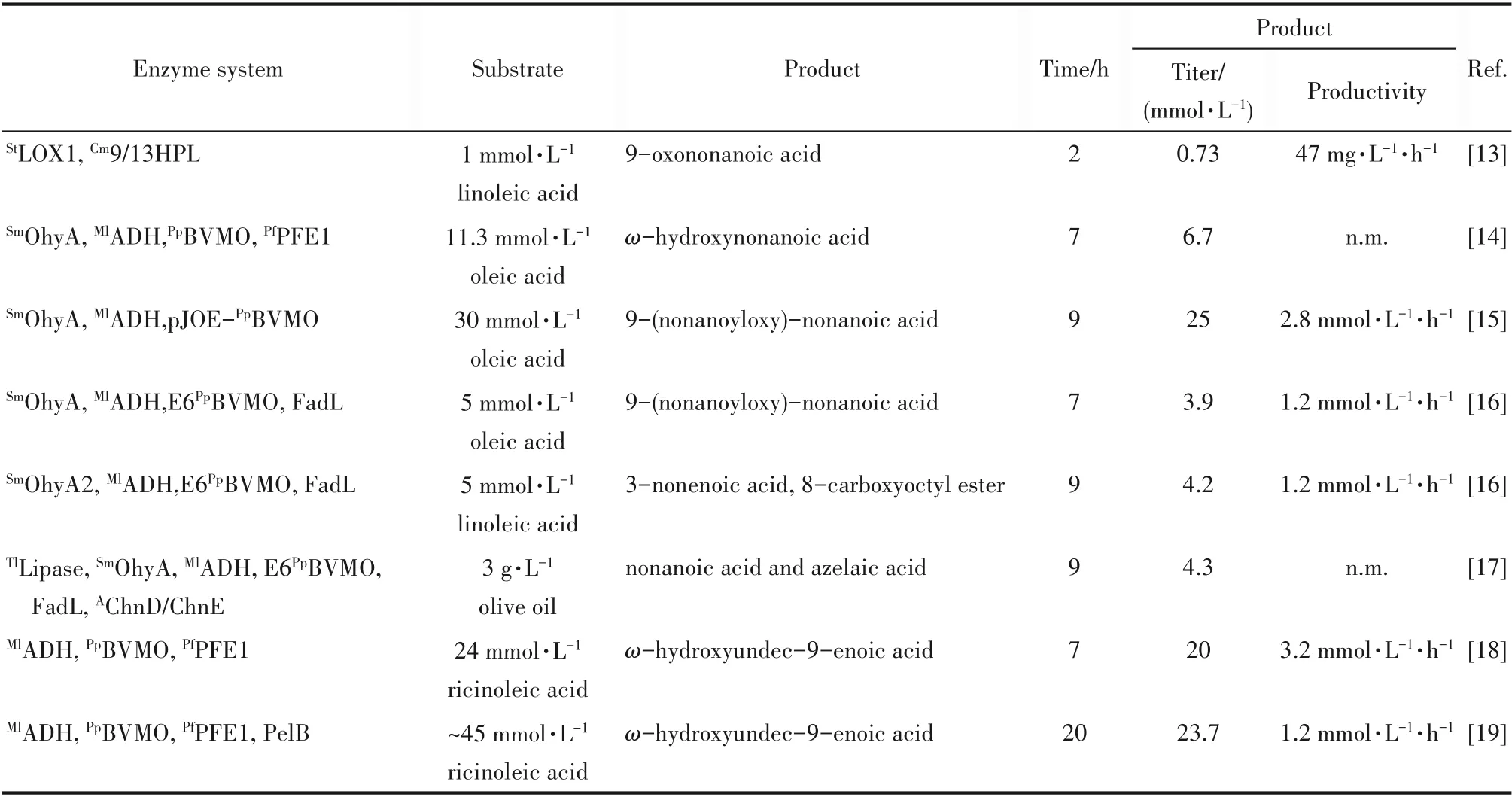
表1 植物油转化生成ω-羟基脂肪酸的多酶级联反应Table 1 Multienzyme cascades to ω-HFAs from plant oils
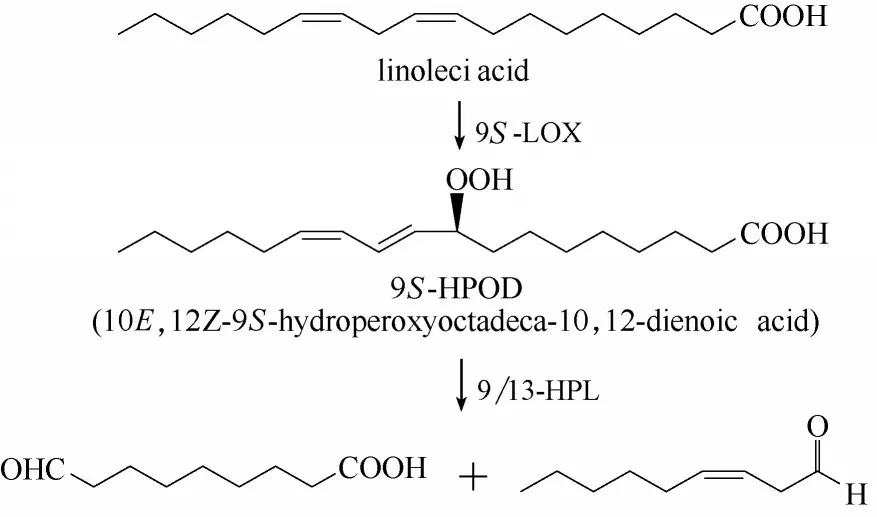
图2 生物催化亚油酸生成9−醛基壬酸Fig.2 Preparation of 9−oxononanoic acid from linoleic acid by LOX and HPL
拜耳−维利格单加氧酶(Baeyer−Villiger monooxygenases, BVMOs)可以活化C—C 键进行扩环,在酮羰基与邻近烃基之间引入一个氧原子而得到相应的酯,并且具有出色的位置和立体选择性,因而被广泛应用于多种酮类化合物的氧化反应中[21]。2013 年,Song 等[14]将来源 于Stenotrophomonas maltophilia 的油酸水合酶(oleate hydratase,OhyA)、来源于M. luteus 的醇脱氢酶ADH 以及P. putida KT2440 的单加氧酶PpBVMO 进行基因共表达,实现油酸向中间酯的转化,再通过P. fluorescens 酯酶的水解,生成正壬酸以及ω−羟基壬酸(图3)。以橄榄油中提取的油酸为底物(11.3 mmol·L−1),最终获得了6.7 mmol·L−1ω−羟基壬酸,得率为60%。然而,用来源于P.fluorescens DSM 50106 的“非常规”PfBVMO 替代PpBVMO 进行反应时,生成的产物完全不同;由于PfBVMO在羰基的另一侧引入氧原子,导致最终的产物为正辛酸以及α,ω−癸二酸。这两个区域选择性恰好相反的BVMO 均由Bornscheuer 等[22−23]最先发现,采用不同选择性的BVMOs有利于丰富脂肪酸催化途径产物的多样性。2018 年,本课题组[24]通过基因挖掘和功能筛选得到来源于铜绿假单胞菌Pseudomonas aeruginosa 的PaBVMO,该酶对碳原子数在17~20 的长链酮酸(10−羰基十七烷酸、9−羰基十八烷酸、10−羰基十八烷酸、10−羰基十九烷酸、10−羰基二十烷酸)的区域选择性均大于90/10 (非常规酯酸/常规酯酸,摩尔比),显著高于所用探针酶PfBVMO,表现出较高的“非常规”区域选择性,更加适合植物油的生物转化路径,可以显著减少副产物,提高底物利用率。
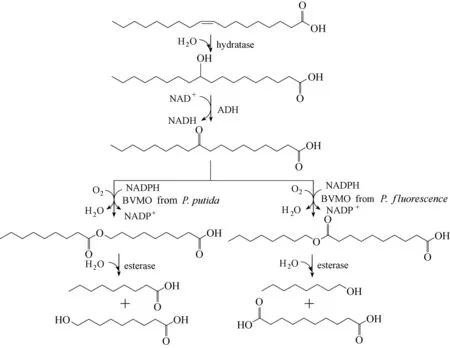
图3 油酸的两条不同生物转化途径Fig.3 Different biotransformation pathways for oleic acid
2016 年,Koppireddi 等[15]采 用 油 酸 水 合 酶(OhyA)、醇脱氢酶ADH 以及单加氧酶PpBVMO 实现油酸向酯的转化后,用化学法对酯进行水解,而不用酯酶,因为水解产物正壬酸对细胞具有毒害作用[25]。此外还发现,级联酶在大肠杆菌细胞中的表达水平影响油酸向酯的生物转化速率,因此,将油酸水合酶和单加氧酶分别构建重组细胞,采用双细胞体系进行反应,明显提高了转化率以及反应速率,30 mmol·L−1油酸,9 h 后可转化为25 mmol·L−1酯,酯生成速率达2.8 mmol·L−1·h−1。
同年,Jeon 等[16]为了进一步强化脂肪酸到酯的转化率,在前期研究基础上,进一步采用Seo 等[26]构建的工程化BVMO 酶(E6BVMO),并对位于细胞外膜的一个长链脂肪酸转运蛋白FadL进行过表达,相比于无FadL的双细胞体系,油酸到酯的转化效率提高了4 倍,由5 mmol·L−1油酸可以获得3.9 mmol·L−1酯,酯的生成速率达1.2 mmol·L−1·h−1。增加细胞添加量,底物上载量可以提高到60 mmol·L−1,8 h 可获得42 mmol·L−1酯,酯的 生 成速 率达5.3 mmol·L−1·h−1。最后采用来源于Thermomyces lanuginosus 的脂肪水解酶(TLL)对酯进行水解,获得壬酸以及9−羟基壬酸。之后,在前期研究基础上[14,27],也对亚油酸进行了转化(图4),与油酸转化不同的是,筛选获得一新型脂肪酸水合酶OhyA2,5 mmol·L−1亚油酸可以转 化 为4.2 mmol·L−1酯,酯 生 成 速 率 达1.2 mmol·L−1·h−1。
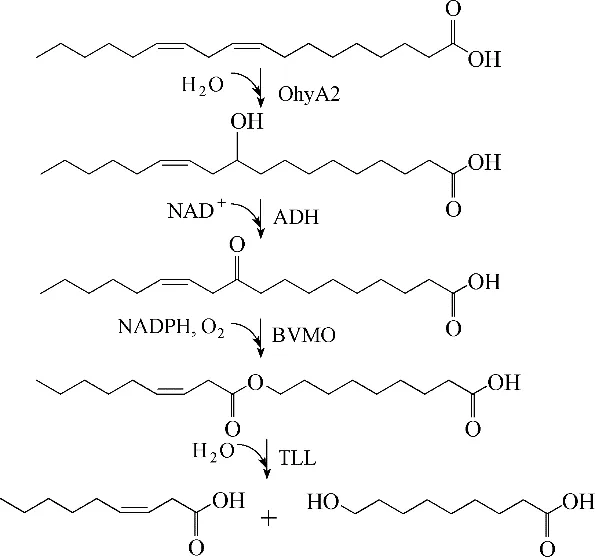
图4 亚油酸的生物转化途径Fig.4 Biotransformation pathway for linoleic acid
2018 年,Seo 等[17]为将产物9−羟基壬酸进一步功能化生成α,β−壬二酸,引入对长链脂肪酸具有较高氧化活力、来源于Acinetobacter sp.NCIMB9871 的醇/醛脱氢酶(ChnD/ChnE)[28],对多种植物油(橄榄油、大豆油、微生物油脂)进行了整细胞催化。其中,以市售的橄榄油为底物,首先采用脂肪酶(lipase)对甘油三酯进行水解获得油酸,再采用整细胞催化剂以及脂肪酶进行转化,可获得正壬酸和壬二酸(图5):加入3 g·L−1橄榄油,壬酸以及壬二酸的产物浓度可以达到4.3 mmol·L−1,比产率为3.1 U·g−1CDW。
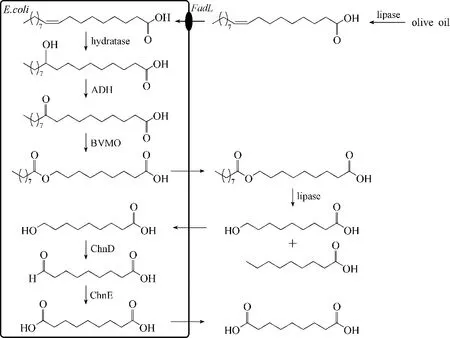
图5 以整细胞及酶催化可再生油脂生物转化的反应途径示意图Fig.5 Schematic diagram of the enzyme/whole−cell biotransformation system of renewable oils
1.2 不饱和ω-羟基脂肪酸的合成
在油酸与亚油酸的生物转化中,主要生成的是壬酸以及9−羟基壬酸;若使用“非常规”BVMO,也会生成辛酸和癸二酸。而以蓖麻油酸为底物时,Song等[14]首次将蓖麻油酸转化生成正庚酸以及ω−羟基−9−十 一 烯 酸(ω −hydroxyundec−9−enoic acid, ω −HUA)。Jang 等[18]采用来源于M. luteus 的醇脱氢酶、P. putida KT2440 的单加氧酶PpBVMO 以及来源于Pseudomonas fluorescens SIK WI 的酯酶,实现蓖麻油酸向庚酸以及ω−HUA 的高效转化(图6):24 mmol·L−1蓖麻油酸,以3.2 mmol·L−1·h−1的速率最终生成20 mmol·L−1正庚酸以及ω−HUA,转化率>80%,产物正庚酸和ω−HUA 的产量分别为2.5 g·L−1和4.0 g·L−1。由于观察到还有中间体酯未完全水解,Cho 等[19]推测可能是由于酯的强疏水性使得其难于被在胞质空间表达的酯酶进行高效催化,所以将酯酶与一个信号肽PelB[29]进行融合表达,使其能被该疏水肽运输到周质空间中,并去除该序列中信号肽酶的识别位点,使酯酶锚定在胞质膜上进行催化,虽然融合蛋白的酶活性有所下降,但是产物ω−HUA 的生成速率以及产物得率均有效提高,产量可达4.7 g·L−1。
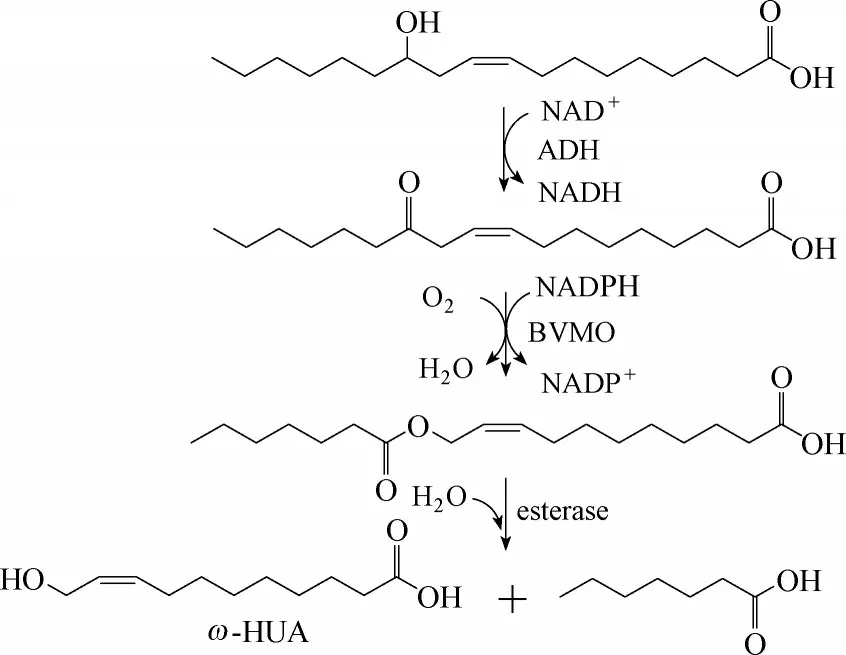
图6 蓖麻油酸的生物转化途径Fig.6 Biotransformation pathway for ricinoleic acid
由于壬酸等脂肪酸产物会破坏细胞的电子传递链和氧化磷酸化[30],且过量的酸在细胞体内会造成酸化,影响胞内pH,从而对细胞的生理和代谢造成影响。为实现植物油生物转化过程的工业应用,以及高生产率、高产物浓度的目标,减少产物对细胞的毒性影响至关重要。这可以通过基因工程、代谢工程对细胞进行改造,Woo 等[31]基于GadA/B 的谷氨酸依赖性耐酸(GDAR)系统,该系统通过催化谷氨酸的脱羧反应来清除细胞内的质子,而在大肠杆菌BL21(DE3)中不活跃,因此通过过表达rcsB和dsrA基因来激活GDAR 系统,从而提高了细胞对正庚酸的耐酸能力;也可以设计水−有机溶剂双相反应体系,使易溶于有机溶剂的有毒产物与细胞实现有效的隔离[32],或在反应中使用吸附树脂,对疏水产物进行原位回收[33],从而降低产物对细胞催化活性的影响。
对于整细胞催化而言,脂肪酸的跨膜转运效率成为一个重要的限速因素。脂肪酸特别是长链脂肪酸需要通过长链脂肪酸转运蛋白FadL 穿过细胞外膜,然后被膜结合的脂肪酸酰基辅酶A 合成酶激活之后,释放到细胞溶胶中[28,34]。Jeon 等[35]也通过研究,发现FadL表达水平是决定长链脂肪酸和羟基脂肪酸整细胞生物转化率的关键因素之一,FadL 的过表达可以提高细胞对脂肪酸的转化速率,Shin 等[36]通过在大肠杆菌中过量表达微囊蛋白Caveolin−1,提高细胞对疏水性底物的内吞作用而提高脂肪酸的转运速率。此外,从上述案例中可以发现,BVMO单加氧酶是脂肪酸转化途径中的一个限速酶,而其在氧化和热刺激下的不稳定性,以及有限的活力仍然是实现生物催化剂工业应用中的一个主要障碍。随着蛋白质工程以及酶工程等领域的发展,可在基因水平上对BVMO 进行分子改造,例如通过密码子优化、融合表达可溶性标签蛋白、分子改造提高酶的比活力以及在转录和翻译水平优化基因的表达等各种手段来增强酶的表达水平和结构稳定性[26,37−38]。
2 ω−氨基脂肪酸
ω−氨基脂肪酸(ω−AmFAs)是重要的一类脂肪酸衍生物,具有α−羧基和ω−氨基双官能团,可以进一步缩合生成聚酰胺聚合体,是重要的聚酰胺合成单体,例如目前广泛应用的6−氨基己酸、ω−氨基十二烷酸等。聚酰胺即尼龙(nylon,polyamide),它是含有酰胺基团(—CO—NH—)的高聚物总称,是目前工业中应用广泛的工程塑料。此外,由于具有良好的综合性能,包括力学性能、耐热性、耐磨损性、耐化学药品性和自润滑性等,且摩擦系数低,有一定的阻燃性,易于加工[39],被广泛用于制作机械、化工、电器等零件。

图7 ω−氨基脂肪酸的化学[41−42]和酶法合成路线Fig.7 Chemical and enzymatic synthesis routes to ω−AmFAs
ω−AmFAs除了作为聚酰胺的重要合成单体外,也具有一定的医疗方面的应用。例如,8−氨基辛酸盐或其氨基衍生物的单体或组合体作为维生素B12的吸收促进剂而研制成口服制剂,可用于预防或治疗维生素B12缺乏症[40]。此外,在一些抗菌药物的合成中,为了调节酯类化合物的活性、毒性和亲脂性,常以ω−AmFAs为基础插入烷基,更好地促进药物的活性并降低对肠胃的毒性[9]。
2.1 ω-氨基脂肪酸的合成
ω−AmFAs由于其广泛的应用价值,其合成方法一直受到化学家和生物工程师的关注。如图7 所示,是ω−AmFAs 合成中常用的化学和生物催化路径。工业上常采用的化学路线是臭氧以及金属镍催化的氧化还原反应,或是烯酸与丙烯腈的交叉复分解反应[43]。在传统的工业制备中,还原胺化工艺常使用非均相催化剂,如二氧化硅或氧化铝负载金属催化剂,并在150~210℃的高温和18~200 bar(1 bar=105Pa)的压力下进行反应[44]。化学合成法的主要优点[45]之一是高时空产率,通常具有较高的反应速率,所得产物只需经过简单的蒸馏等下游加工,即可获得较高的分离得率,提高了时空产率。此外,使用的多相催化剂易于循环再用,并使用廉价的还原剂即氢气和氨气,显著降低了成本。然而,由于反应需要高温等条件,因此需要较大的能源消耗,较高的反应温度难以避免一些副产物的形成,例如仲胺和叔胺等,反应选择性有限。
以脂肪酸为原材料,生产ω−AmFAs的工业应用仍较少[46]。对脂肪酸烷基链上的惰性C—H 键进行官能团化是化学工业上的一个难点,而随着生物催化的发展,生物发酵以及酶催化(如单加氧酶P450)可以实现这一转化。Arkema France[46]通过发酵工程将油酸转化为二酸,经臭氧分解后被还原得到9−醛基壬酸,在Ra−Ni 或者Pd/C[47]的催化下,使用NH3/H2还原胺化生成9−氨基壬酸,如图8所示。
近年来,随着生物技术研究的发展,越来越多的酶催化被应用于ω−AmFAs 的生产研究。根据起始原料的不同,ω−AmFAs 的生物合成途径有所不同,如图7 所示:(1)从游离脂肪酸出发,通过P450 羟化酶在末端羟化形成ω−HFAs[48];(2)从脂肪酸酯[27]或内酯[49]出发,用酯酶进行水解生成ω−HFAs,通过醇脱氢酶氧化生成ω−醛基脂肪酸,再用转氨酶或胺脱氢酶将醛酸胺化生成ω−AmFAs。
对于脂肪酸的生物胺化反应,过去的研究主要集中于低分子量的短链脂肪酸如乳酸、琥珀酸等[50]。对于长链脂肪酸,可以直接胺化植物油生成如10−氨基硬脂酸[51]。然而,以中链脂肪酸衍生物为单体合成的一些生物塑料表现出与聚乙烯和其他生物塑料相似甚至更优的物理化学性质,但其生物合成目前仍是一项挑战性的任务[27]。因此本节主要介绍近年来中链ω−AmFAs的生物合成研究进展。
2.2 多酶级联催化合成ω-氨基脂肪酸
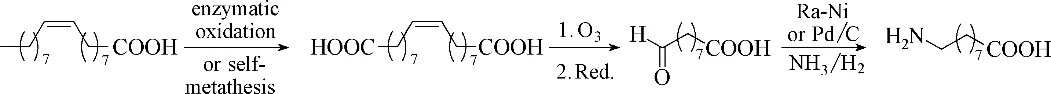
图8 化学/酶法合成9−氨基壬酸Fig.8 Chemical/enzymatic synthesis of 9−aminonanoic acid

表2 多酶级联合成不同链长的ω-氨基脂肪酸的路线Table 2 Multi-enzymatic synthesis of ω-AmFAs in recombinant E.coli
从上述介绍可以发现,ω−AmFAs的生物合成涉及多个反应,需要多个不同的酶参与。为简化反应步骤,实现ω−AmFAs 的一步合成,越来越多的生物化学家致力于研究多酶级联反应。目前,根据合成底物的不同,ω−AmFAs的多酶级联合成可以分为两类:一类是从内酯或ω−HFAs 生成ω−AmFAs 的多酶级联,不需要单加氧酶的参与;另一类是从脂肪酸转化为ω−AmFAs的多酶级联,需要一个可以在脂肪酸碳链末端进行羟化的单加氧酶。表2 对近年来ω−AmFAs的合成研究进行了汇总。
2.2.1 由内酯或ω−羟基脂肪酸生成ω−氨基脂肪酸的多酶级联 从脂肪酸酯出发,通过酯酶水解生成ω−HFAs,对于一些特殊链长的氨基脂肪酸,如6−氨基己酸,也可以通过环内酯的水解获得ω−HFAs,再通过醇脱氢酶和转氨酶催化生成ω−AmFAs。2014年,Sattler 等[49]设计了脱氢与转氨两步反应,并引入丙氨酸脱氢酶实现辅因子自给自足的生物催化级联,使得己内酯成功转化为6−氨基己酸(图9)。为最小化末端6−羟基己酸的形成,将己内酯在甲醇存在下,在水溶液中转化为相应的酯,以保护羧基,并首次发现马肝酯酶对己内酯环的催化效率最高,最终50 mmol·L−1己内酯反应20 h后,产物6−氨基己酸的占比达75%。
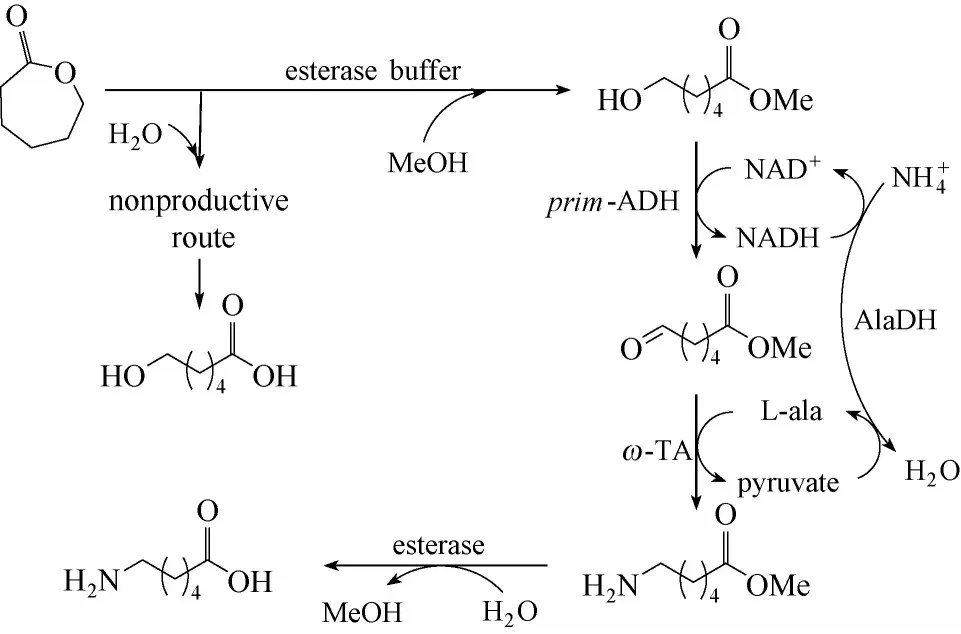
图9 由己内酯合成6−氨基己酸的催化路线设计Fig.9 Catalytic route of caprolactone to 6−aminocaproic acid
2014 年,以12−羟基硬脂酸为底物,Song 等[27]采用一锅两步级联催化生成ω−氨基十一烷酸,先采用来自Micrococcus luteus NCTC2665 的醇脱氢酶ADH、来自P. putida KT2440 的单加氧酶BVMO 以及来自P. fluorescens SIK WI 的酯酶将12−羟基硬脂酸转化为正庚酸以及11−羟基脂肪酸,之后加入来源于P.putida GPo1的醇脱氢酶AlkJ以及来自S.pomeroyi的转氨酶,进一步将11−羟基十一烷酸转化为11−氨基十一烷酸(图10)。5 mmol·L−1底物12−羟基硬脂酸,最终转化为2.5 mmol·L−111−氨基十一烷酸,而分离得率只有36%,这是由于AlkJ 会进一步过氧化而生成少量(0.7 mmol·L−1)的副产物十一烷二酸。
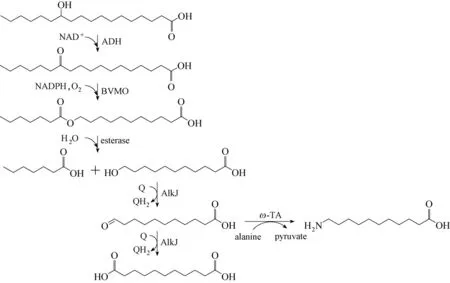
图10 多酶级联转化12−羟基硬脂酸生成11−氨基十一烷酸Fig.10 Biotransformation pathway of 12−hydroxystearic acid into 11−aminoundecanoic acid
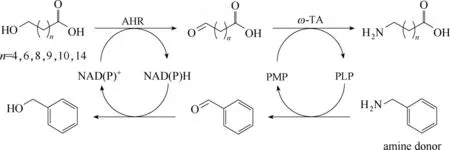
图11 生成ω−氨基脂肪酸的反向平行级联设计Fig.11 Design of two interconnected two−step sequences,parallel anti−sense cascade,to produce ω−AmFAs
使用同一转氨酶,2018 年,Sung 等[52]将其与来自Synechocystis sp. slr1192 的醛还原酶(AHR)偶联,并以氨基供体苄胺为辅底物,设计了涉及两步反应的平行级联[56],成功将不同链长(C6~C16)的ω−HFAs转化为相应的ω−AmFAs,同时生成副产物苯甲醇(图11),采用共表达的整细胞进行催化反应,在100 mmol·L−1底物上载量下,转化率可达到80%~95%。
2.2.2 脂肪酸转化为ω−氨基脂肪酸的多酶级联从脂肪酸出发合成ω−AmFAs,则需要加氧酶和醇脱氢酶进行末端加氧以及脱氢得到醛酸,才能进一步胺化生成氨基酸,而加氧酶往往是整条路线中的限速酶。目前,烷烃的选择性末端羟化仍然是一项具有挑战性的任务[57],中链脂肪酸末端羟化反应主要用到的是细胞色素P450 单加氧酶(如CYP153)[58]以及烷烃氧化酶AlkB[59]。烷烃氧化酶体系AlkBGT 是由三个酶构成的,单加氧酶AlkB以及两个负责电子传递的辅助蛋白AlkG 和AlkT[59]。2011 年,Schrewe等[60]将AlkBGT 在大肠杆菌中异源表达,对不同链长(C5~C12)的脂肪酸甲酯进行了研究,其中对壬酸甲酯的末端羟化活性最高,达到104 U·g−1CDW。在此基础上,将该单加氧酶体系与来自Chromobacterium violaceum 的转氨酶CV2025相结合[53],通过重组整细胞反应实现了十二烷酸甲酯的末端胺化(图12),2.9 mmol·L−1底 物 十 二 烷 酸 甲 酯,90 min 后 生 成0.13 mmol·L−1ω−氨基十二烷酸甲酯,最大的胺形成速率仅1.5 μmol·min−1·g−1CDW。由于单加氧酶AlkBGT导致了中间产物醛的过氧化,生成了相同产物浓度的副产物酸,且底物水溶性差也是导致转化率低的重要原因。此外,认为可以进一步通过催化剂工程来提高疏水性底物的可获得性,并应用双相反应体系来解决该反应中底物或产物对酶的抑制作用。
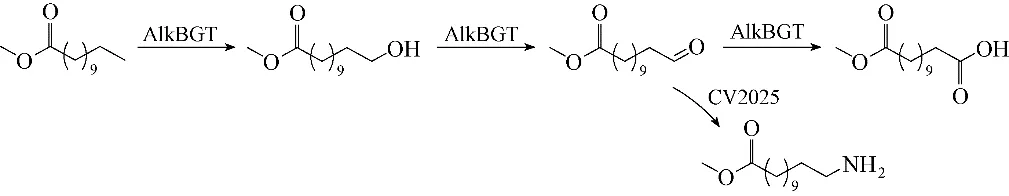
图12 十二烷酸甲酯的末端氨基功能化Fig.12 Amination of ω−functionalized methyl dodecanoate
为了避免AlkB 可逆反应中中间产物的积累,Kirmair 等[61]使用了与AlkB 同一来源的醇脱氢酶AlkJ,并进行了酶学性质的研究,该酶能不可逆催化醇氧化生成醛,然而,在反应中也同样会过氧化催化生成副产物。2016年,Ladkau等[54]在上述Schrewe的AlkBGT 体系催化研究基础上,引入外膜蛋白AlkL,提高了细胞对疏水性底物的摄取以及AlkBGT的氧化活力,转氨酶活力提高了7.3 倍,达11 U·g−1CDW,生成0.55 mmol·L−1ω−氨基十二烷酸甲酯(基于2.9 mmol·L−1底物十二烷酸甲酯)。为减少外源添加高浓度L−丙氨酸的成本,通过借鉴Sattler 的研究[49],引入了丙氨酸脱氢酶AlaDH 实现L−丙氨酸的胞内循环,同时引入催化不可逆反应的醇脱氢酶AlkJ,明显增加了最终产物ω−氨基十二烷酸甲酯的浓度。
除了AlkBGT 单加氧酶体系,细胞色素P450 中的CYP153 亚家族也具有末端羟化反应的催化能力,并具有较高的末端区域选择性。2018 年,Ahsan等[48]发现了一新型单加氧酶,来自Mycobacterium parascrofulaceum 的CYP153A (MprCYP153A),采 用CamAB 为氧化还原伴侣,对十二氨基烷酸具有较高的转化率(1 mmol·L−1底物,转化率>99%)。此外,将醇脱氢酶AlkJ 与来自M. loti 的转氨酶mll 1207 ω−TA 进行共表达后,将MprCYP153A/CamAB 与AlkJ/mll 1207 ω−TA 双细胞进行一锅反应[图13(a)],苯乙胺为氨基供体,催化2 mmol·L−1十二烷酸,6 h 后生成0.6 mmol·L−1ω −氨 基 十 二 烷 酸。为 了 简 化CYP153A 的羟化反应,该课题组[55]将来自Alcanivorax borkumensis SK2的CYP153A13与天然自给自足的细胞色素P450 还原酶(CPR)进行融合表达,构建了自给自足型人工P450酶(CYP153A13BM3CPR),将该酶与醇脱氢酶AlkJ 及转氨酶Sp ω−TA 一起共表达后,可以实现十二烷酸到ω−氨基十二烷酸的整细胞催化[图13(b)]。通过优化反应体系,反应24 h后,图13(b)体系可以从10 mmol·L−1十二烷酸中获得1.48 mmol·L−1ω−氨基十二烷酸,产物浓度相比于图13(a)体系提高了5倍。
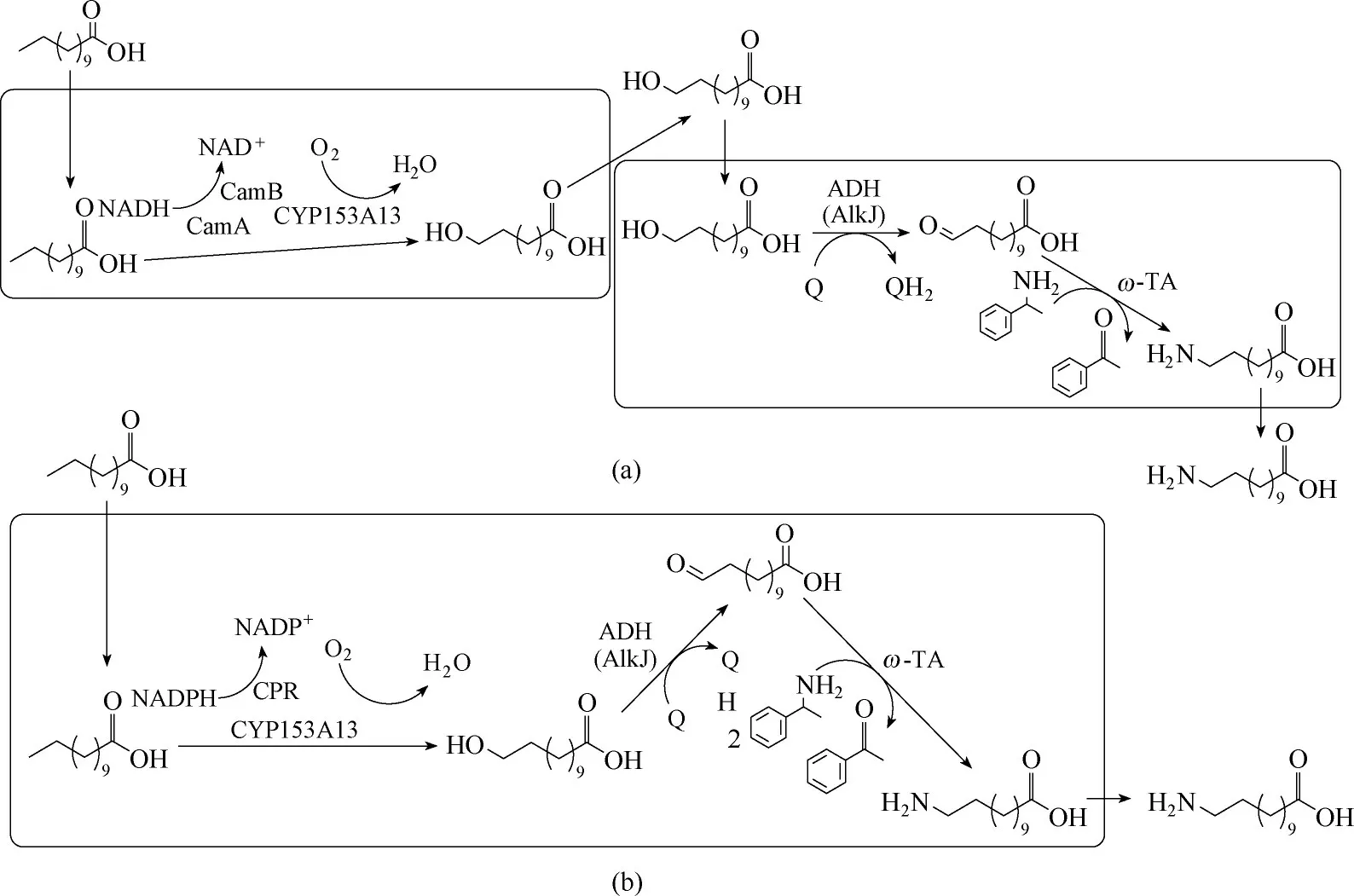
图13 ω−氨基十二烷酸的整细胞合成Fig.13 The biosynthesis of ω−amino dodecanoic acid(ω−AmDDA)from dodecanoic acid(DDA)
从上述案例中可以发现,ω−醛基脂肪酸的胺化反应主要采用了ω−转氨酶进行催化。转氨酶以5−磷酸吡哆醛(PLP)为辅因子,能可逆催化羰基与氨基之间的氨基转移反应[62]。然而,转氨酶反应中的热力学平衡问题影响了转氨酶的催化效率[63]。为推动反应平衡向产物形成方向移动,可以通过反应工程手段来实现[64],例如加入大幅过量的氨基供体(如异丙胺);原位移除产物或副产物;或如图9 偶联其他酶促反应,使反应副产物转化为原始的辅底物加以循环利用,或生成其他易于移除的成分[65]。因此,在转氨酶的应用中,选择合适的酶并应用一个合适的推动平衡策略至关重要。
近日,Mutti 课题组[66]报道了一株由赖氨酸脱氢酶改造而来的胺脱氢酶(AmDH),对来源于G.stearothermophilus 菌株的野生型赖氨酸脱氢酶进行计算机模拟并基于其结构信息进行分子改造,获得的最佳变体LEAmDH−V1 (F173A)不仅具有较好的热稳定性(Tm= 69℃),并且可以催化己醛酸生成6−氨基己酸,实现50 mmol·L−1己醛酸的完全胺化(转化率>99%)。此外,2019 年,Citoler 等[67]将羧酸还原酶(carboxylic acid reductases, CARs)与转氨酶级联,对C6~C18的饱和或不饱和脂肪酸进行酶法胺化,将脂肪酸的羧基还原为醛基后,胺化生成脂肪胺。此外值得一提的是,人们发现真菌中存在一类非特异性过氧化物酶(unspecific peroxygenases,UPOs)[68],与细胞色素P450单加氧酶的功能类似,对脂肪酸的惰性碳链也具有选择性氧化的能力,目前已经实现了来源于Marasmius rotula 的UPO 酶基因在大肠杆菌中的异源过量表达[69],这为中长链脂肪酸的生物羟化及胺化反应提供了新的路径。
3 总结与展望
从植物油或其脂肪酸出发,通过非天然的微生物酶促转化反应,可以生产高附加值的ω−HFAs 与ω−AmFAs,可用于生产化妆品、塑料、工业材料等,具有光明的应用研究和开发前景。在中链ω−HFAs与ω−AmFAs 的研究中,主要应用了油酸水合酶、醇脱氢酶、BVMO 单加氧酶、酯酶、P450 羟化酶以及转氨酶等催化元件,然而目前大多数的生物催化反应仍停留在实验室研究阶段,还无法实现工业化规模生产,这主要是由于酶的稳定性不足以及催化活力还较低,尤其是BVMO 单加氧酶和P450羟化酶成为主要的限速瓶颈。此外,大肠杆菌的低耐受性、底物的强疏水性和产物对细胞的高毒性,也是目前底物上载量低、生产强度较低的主要原因。
从上述案例可以发现,ω−HFAs 与ω−AmFAs 的生物催化合成普遍应用了多酶级联催化的生物反应过程,且采用整细胞或者粗酶液在一个反应器内进行体内或体外级联反应,这有助于实现辅因子的有效自我循环,并避免了中间体的分离和纯化,跳过了烦琐耗时的中间步骤,避免了中间产物的积累,可以促使可逆反应的完成[70],以及最大限度地减少了用于萃取和纯化的有机溶剂用量,具有较高的原子经济性和较低的环境影响因子[71],工业化应用潜力巨大。然而,多酶级联反应涉及多个反应与酶,相较于单步反应更为复杂,在实际应用中需要考虑多种因素。多种异源酶之间的相互协调以及反应条件相互兼容是多酶级联中的关键问题,也是一个颇具挑战性的难题[72]。对多酶共表达的整细胞体系,如何调节活性酶的表达比例尤为重要,目前可用的手段是通过使用拷贝数不同的载体,或者优化启动子强度和核糖体结合位点(RBS)等技术,实现多个酶基因的协调可控表达[73]。
因此,为了提高ω−HFAs 与ω−AmFAs 作为工业产品的潜力,人们仍需要注重提高级联酶的稳定性以及催化效率,提高微生物细胞的耐受性,对限速酶在解析其结构与功能关系的基础上进行分子改造,以克服脂肪酸生物转化中的瓶颈问题;并且在多酶级联反应体系中,就如何减少中间产物的积累,加快物料的传递和控制平衡问题,需要根据反应类型以及产品的不同,进行系统性工程优化和应用研究,需要考虑包括生物转化途径的设计、级联酶催化元件的筛选以及分子工程改造,多酶表达之间的平衡协调以及系统工程等各方面的因素[74],构建一个高效的工程化整细胞生物催化剂,以实现大规模的工业化应用。
