灯盏乙素对β淀粉样蛋白诱导的神经母细胞瘤细胞谷氨酸/γ氨基丁酸通路的影响
2020-09-28李晓宇于燕妮郭莉莉
李晓宇,于燕妮,郭莉莉
1贵州医科大学,贵阳550001;2贵阳市第一人民医院
阿尔茨海默病(AD)是一种慢性进行性神经退行性疾病,涉及记忆障碍和行为障碍。β淀粉样蛋白(Aβ)级联假说是AD发生发展的中心环节,Aβ可导致细胞外谷氨酸(Glu)水平升高,从而产生Glu兴奋性神经毒性损伤神经细胞。而细胞外Glu浓度及作用主要与Glu/γ氨基丁酸(GABA)对神经系统的平衡调节作用有关[1,2]。Glu转运体与GABA转运体协同作用,共同维持突触间隙的Glu和GABA含量,参与正常神经元活动及大脑的多种病理过程;转运功能一旦受损或协同失衡,将导致突触间隙内的Glu浓度升高,持续激活Glu受体,继之造成突触后神经元持续兴奋,介导细胞Ca2+超载,产生大量自由基,形成氧化应激,致神经元损伤,产生神经毒性[3]。EAAT3是Glu通路的重要转运蛋白,N-甲基-D-天门冬氨酸受体亚型1(NMDAR1)是Glu通路的重要受体,而γ氨基丁酸转运体1(GAT-1)是GABA通路的重要转运蛋白,三者共同参与介导Glu兴奋性神经毒性过程。灯盏乙素(Scu)是灯盏细辛的药理活性成分,其通过抗氧化、清除ROS、抗炎、抑制细胞凋亡等途径发挥神经保护作用[4]。2019年2~10月,我们将Aβ1-42作用于人源性神经母细胞瘤SH-SY5Y细胞,诱导AD神经元损伤细胞模型,观察Scu对细胞中Glu/GABA平衡系统协同作用的相关递质、蛋白以及氧化应激指标的影响,探讨Scu抑制Glu兴奋性神经毒性损伤的机制。
1 材料与方法
1.1 材料
1.1.1 细胞与药物 人源性神经母细胞瘤(SH-SY5Y)细胞株,购自中国科学院昆明细胞生物研究所。实验用Scu(南京泽朗医药科技有限公司,纯度98%,批号:ZL20161012037),精确称量分装后置4 ℃保存,使用前加入DMEM充分稀释,并用0.22 μm微孔注射滤器过滤。Aβ1-42寡聚体粉末(强耀生物科技有限公司,纯度95.27%,货号:04010011526),用预冷的六氟异丙醇(HFIP)配制成1 mmol/L的溶液,风干,-80 ℃保存,使用前加不含酚红的F12培养基充分溶解,4 ℃放置24 h后离心取上清。
1.1.2 试剂与仪器 Glu、GABA、总超氧化物歧化酶(T-SOD)、丙二醛(MDA)检测试剂盒(南京建成公司);EAAT3、GAT-1、NMDAR1抗体(博士德公司);多功能酶标仪(美国Thermo Fisher公司);台式高速冷冻离心机(Eppendorf Germany公司);BIO-RAD电泳设备(美国Bio-Rad公司)。
1.2 实验方法
1.2.1 细胞培养 取SH-SY5Y细胞,加入完全培养液(90%高糖DMEM,10%胎牛血清,1%青/链霉素),置于5% CO2、37 ℃培养箱中培养,每2 d换液1次,3~4 d传代。实验重复3次。
1.2.2 细胞分组与处理 取对数生长期细胞,分为对照组、Aβ组、Scu组、Scu+Aβ组。对照组常规培养,不加任何药物;Scu组加入10 μmol/L Scu作用48 h;Aβ组加入1 μmol/L Aβ作用24 h;Scu+Aβ组先加入10 μmol/L Scu预处理24 h,再加入1 μmol/L Aβ作用24 h。
1.2.3 细胞外液Glu含量检测 采用生物化学法。收集0.2 mL细胞培养液,加入0.6 mL试剂一充分混匀,3 000 r/min离心10 min,取上清。设定空白管、标准管、测定管,分别加入0.5 mL蒸馏水、Glu标准应用液、待测上清,然后每管依次加入试剂二、试剂三、试剂四,混匀,上酶标仪于340 nm处测吸光度A1。各管再加入试剂五,混匀,37 ℃水浴40 min,于340 nm处测吸光度A2,计算Glu浓度。Glu浓度=[(测定A2-测定A1)-(空白A2-空白A1)]/[(标准A2-标准A1)-(空白A2-空白A1)]×标准品浓度×样本稀释倍数。
1.2.4 细胞外液GABA含量检测 采用ELISA法。收集各组细胞培养液,3 000 r/min离心10 min,取上清。取96孔板,设置空白孔、标准孔、待测样本孔,加入样本和标准品;加入生物素抗原,37 ℃孵育30 min,洗板5次;加亲和素HRP,37 ℃孵育30 min,洗板5次;加显色液A、B,37 ℃显色10 min;加终止液。上酶标仪于450 nm处测A值。根据标准品浓度及A值绘制标准曲线,根据标准曲线回归方程计算GABA含量。
1.2.5 EAAT3、GAT-1、NMDAR1蛋白表达检测 采用Western blotting法。收集各组细胞,加入蛋白裂解液提取总蛋白,进行蛋白定量。采用SDS-聚丙烯酰胺凝胶电泳,转至PVDF膜,分别加入相应的蛋白一抗(β-actin稀释比例1∶200;EAAT3稀释比例1∶200;GAT-1稀释比例1∶200;NMDAR1稀释比例1∶500)和二抗(二抗稀释比例1∶50 000),孵育后曝光显影,定影。用Bandscan分析灰度值,以目标条带与β-actin条带的灰度比值表示目标蛋白的相对表达量。
1.2.6 细胞T-SOD活力检测 采用黄嘌呤氧化酶法。收集各组细胞,用反复冻融的方法破碎细胞,离心,取上清,用BCA法检测上清液中的蛋白浓度,取96孔板,设定对照孔(蒸馏水)和测定孔(细胞上清),依次加入工作液后混匀,37 ℃水浴40 min。加入显色剂混匀后室温放置10 min,于波长550 nm处测A值,根据公式计算T-SOD活力。总SOD活力=(对照孔A值-测定孔A值)/对照孔A值/0.5×(反应液总体积/取样量)/蛋白浓度。
1.2.7 细胞MDA含量检测 采用TBA法。收集各组细胞,用反复冻融的方法破碎细胞,离心,取上清,用BCA法检测上清液中的蛋白浓度。取5 mL试管,设置空白管、标准管、测定管、对照管。依次加入工作液后混匀,95 ℃水浴40 min,取出后流水冷却,4 000 r/min离心10 min。取上清,于532 nm处测A值,并计算MDA浓度。MDA浓度=[(测定管A值-对照管A值)/(标准管A值-空白管A值)]×标准品浓度/待测样本蛋白浓度。
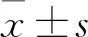
2 结果
2.1 各组细胞外液Glu、GABA含量比较 与对照组比较,Scu组细胞外液Glu、GABA无明显变化(P均>0.05),Aβ组细胞外液Glu升高、GABA降低(P均<0.05);与Aβ组比较,Scu+Aβ组细胞外液Glu降低、GABA升高(P均<0.05)。见表1。

表1 各组细胞外液Glu、GABA含量比较
2.2 各组细胞EAAT3、GAT-1、NMDAR1蛋白表达比较 与对照组比较,Aβ组细胞EAAT3、GAT-1和NMDAR1蛋白表达升高(P均<0.05);与Aβ组比较,Scu+Aβ组细胞EAAT3、GAT-1和NMDAR1蛋白表达降低(P均<0.05)。见表2。
表2 各组细胞GAT-1、EAAT3、NMDAR1蛋白相对表达量比较
2.3 各组细胞T-SOD活力、MDA含量比较 与对照组比较,Aβ组细胞T-SOD活力降低、MDA含量升高(P均<0.05);与Aβ组比较,Scu+Aβ组细胞T-SOD活力升高、MDA含量降低(P均<0.05)。见表3。
表3 各组细胞T-SOD活力、MDA含量比较
3 讨论
Scu是从中草药灯盏花中提取的黄酮类有效成分,既往多用于心脑血管疾病的治疗。近年研究发现,Scu还有保护神经细胞的作用。我们的前期研究显示,Scu能够介导K+-ATP通道开放、调控IP3R-Ca2+通道,通过调控组蛋白H2A、H2B、H3表达来抑制Aβ的神经毒性[5~7]。本研究进一步观察Scu对Aβ诱导的神经母细胞瘤细胞Glu/GABA通路的影响。
Glu是大脑最主要的兴奋性神经递质。Glu作用于突触后膜的离子型谷氨酸受体产生生理作用,参与神经元通信、突触可塑性、神经元生长、分化以及学习和记忆等生理过程[8]。NMDAR属于离子型谷氨酸受体的亚型之一,NMDAR1是NMDAR的功能性亚单位,是Glu通路的重要受体。当Glu完成生理活动后,突触间隙并没有相应的酶水解Glu失活,必须由兴奋性氨基酸转运体(EAAT)转运清除,维持突触间隙Glu的低水平。EAAT3是EAAT的亚型之一,主要表达于神经元细胞,参与维持大脑局部神经细胞外的Glu浓度[9]。
GABA是大脑重要的抑制性神经递质。当GABA神经元兴奋时,释放GABA作用于Glu神经元的GABA受体,抑制神经元释放Glu,此过程叫突触前抑制;GABA也可以作用于突触后膜的GABA受体,引起Cl-内流,抑制突触后神经元兴奋,此过程叫突触后抑制。GABA在突触间隙的浓度需要GABA转运体(GAT)来维持。GAT-1是GAT表达最丰富、最重要的亚型,可促进GABA回吸收到突触前神经元中,从而控制突触间隙GABA含量[10]。
GAT-1和EAAT3都是间接耗能的转运蛋白,当供能不足时二者功能发生异常,并且顺浓度梯度释放GABA和Glu[11]。Glu递质通路和GABA递质通路相互拮抗,共同维持神经细胞的兴奋/抑制的平衡。当GABA的抑制作用减弱,或细胞释放Glu过多,或Glu异常转运等情况下细胞外液Glu增加,过度激活离子型Glu受体,由于NMDAR的亲钙性,会引发细胞外Ca2+内流增加,导致细胞内钙超载,激活大量钙离子依赖性酶,如蛋白激酶C、磷酸脂酶A、一氧化氮合酶等,使蛋白质水解、细胞骨架和细胞膜受损等,同时产生大量自由基,氧化与抗氧化失衡,氧化应激形成,致神经元损伤,该过程称为Glu的兴奋性毒性的级联反应[8]。
本研究结果显示,Aβ寡聚体作用于SH-SY5Y细胞后,细胞外液Glu增加、GABA降低。突触间隙Glu、GABA含量主要取决于神经细胞对Glu、GABA的释放和摄取清除功能,提示Aβ寡聚体可能影响Glu、GABA的释放和摄取功能介导细胞外液Glu的增加。体内外研究均显示,Aβ寡聚体能与神经细胞作用促使细胞释放Glu增加[12],Glu作用于突触后膜增加的NMDAR,NMDAR激活后主要介导Ca2+内流,细胞内Ca2+增加。线粒体作为细胞Ca2+稳态的重要调节器,线粒体不仅通过钙单向转运蛋白吸收大量的Ca2+,而且促进ATP依赖性钙的挤出以调节维持细胞内Ca2+的生理浓度[13]。轻度的Glu兴奋性神经毒性介导的细胞内Ca2+增加会导致短暂的线粒体去极化,可逆的ATP合成减少,而持续的兴奋性毒性或者严重的Ca2+稳态失衡会导致不可逆的线粒体去极化、永久性能量崩塌和离子失衡[14]。如果Aβ毒性不解除,线粒体对Aβ诱发的Glu毒性介导的Ca2+稳态失调难以纠正,并且线粒体Ca2+升高,抑制ATP合成[7,13]。提示Aβ毒性可能介导Glu兴奋毒性,并且导致线粒体功能异常。
突触递质的传递过程是高耗能过程,所以尽管本研究观察到EAAT3表达增加,但结果提示细胞外液Glu并没有减少反而增加,提示转运Glu功能异常,可能顺Glu浓度梯度释放细胞内的Glu,更加重了细胞外液Glu的高浓度,而GABA并没有升高,并不是表达增加的GAT-1转运了GABA,因为GAT-1的正常转运功能同样是高耗能过程。研究表明,GABA的合成原料是GABA神经元上的EAAT3转运入细胞的Glu,在谷氨酸脱羧酶催化下参与合成GABA[2],然而EAAT3异常转运Glu,导致GABA神经元利用Glu合成GABA减少,GABA释放减少,提示GABA减少可能与GABA合成异常有关。GABA的抑制作用减弱,加剧兴奋/抑制信号失衡并进一步促进兴奋性毒性,Glu持续作用并强直激活NMDAR介导细胞内Ca2+超载,线粒体基质内大量Ca2+负载,引起不可逆转的线粒体功能障碍,呼吸链异常,氧自由基增加,打破氧化与抗氧化平衡,介导氧化应激,攻击细胞膜结构和核酸[15]。
SOD是生物体内的一种重要的抗氧化剂。MDA是脂质过氧化产物之一,其含量可反映生物膜过氧化的程度,也间接反映导致生物膜脂质过氧化的氧自由基的含量。抗氧化剂SOD减少与自由基增加提示细胞存在氧化应激和线粒体损伤。我们的前期实验显示,Scu能抑制线粒体通透性转换孔(MPTP)的开放,稳定线粒体功能,增加ATP生成,减少氧自由基和凋亡启动因子释放,进而减少氧化应激损伤和凋亡发生[7,13]。本研究结果显示,与对照组比较,Aβ组T-SOD活力降低、MDA含量升高,表明Aβ诱导神经母细胞瘤发生氧化损伤;与Aβ组比较,Scu+Aβ组T-SOD活力升高、MDA含量降低,表明Scu具有抗氧化作用。
研究显示,Scu可通过抗氧化改善线粒体功能,增加ATP生成,从而改善EAAT3、GAT-1的转运功能。EAAT3转运清除细胞外增加的Glu,并促进GABA合成释放,细胞外低浓度的GABA不能触发GAT-1转运功能,因为此时Glu毒性没有完全纠正,突触间隙需要GABA介导抑制信号抑制神经细胞释放Glu。本研究结果显示,Scu+Aβ组细胞外液Glu含量减少,GABA含量增加,提示Scu重新调节了细胞外Glu、GABA平衡,降低了Glu的兴奋性神经毒性。经Scu干预后,Aβ介导的EAAT3、GAT-1、NMDAR1蛋白表达降低。尽管Scu+Aβ组上述指标未达到正常对照组的水平,但较Aβ组有明显改善,并且Scu组与正常对照组所有指标均无统计学差异,说明Scu对正常细胞没有影响,Scu的调节作用可能发生在Aβ诱导Glu/GABA通路改变之后。
综上所述,Scu可能通过抗氧化作用改善线粒体功能障碍,间接改善EAAT3和GAT-1的转运障碍,同时下调谷氨酸受体亚型NMDAR1的表达,减少Glu的作用受体,从而减轻Aβ介导的谷氨酸兴奋性神经毒性损伤。
