纳米级过硫酸铵微胶囊的制备及性能
2020-09-27李晓丹李光辉郑家桢周伟康
李晓丹,李光辉,郑家桢,周伟康
(燕山大学 车辆与能源学院,河北 秦皇岛 066004)
目前,中国70%的油田为储层渗透率低、丰度低、产能低的低渗透油田。其开采难度大、效率低,能否进行高效的开采,对中国石油产量的影响十分重要[1-3]。压裂技术通过在储层注入压裂液,在目标层段形成高压而使其产生裂缝,从而改善了储层的渗透率。因此,压裂成为提高低渗透油田油气产量的重要措施之一,而压裂液的性能是压裂技术的关键。
压裂液主要包括稠化剂、交联剂、高温稳定剂、支撑剂、破胶剂等组分。其中,支撑剂进入压裂裂缝、支撑裂缝,保持裂缝稳定;破胶剂则使压裂液破胶降黏,排出地层;因此,压裂液注入储层,既改善了储层的渗透率,又不污染地层、环境友好。然而,压裂技术是否有效,很大程度上取决于压裂液能否在精准的时间内破胶。压裂液提前破胶,则无法起到造缝作用;滞后破胶或破胶不彻底,则会导致储层污染。因此,压裂液中破胶剂的性能至关重要。
常用的压裂液破胶剂(过硫酸铵、过硫酸钾等),都存在提前破胶或破胶不彻底的问题[4-5],破胶时间和破胶程度难于控制。因此,微胶囊技术被引入至压裂技术领域。微胶囊破胶剂采用包裹技术,在普通破胶剂外覆盖一层高分子聚合物,从而形成微胶囊型破胶剂[6]。早期的微胶囊破胶剂的生产采用机械包覆法[7],即采用空气悬浮法(Wurster流化床法),在破胶剂颗粒外包覆一层高分子材料,得到一种膜层完整均匀、包覆率较高的微胶囊延迟破胶剂。何显儒等[8]采用介质分散法,利用聚合物液态分散体系,通过改变溶剂条件,使聚合物在相分离过程中逐步包裹于芯材上。陈挺等[9]将聚合物单体与过硫酸铵制备成水溶液,采用乳液聚合方法制备了不同囊衣的微胶囊破胶剂。
但这些方法制备的微胶囊的粒径较大(几百微米至几毫米),大于低渗透油田孔喉平均直径(26~43 μm),因而容易堵塞孔隙喉道,影响储层基质的渗透率。因此,应减小微胶囊破胶剂的粒径,制备纳米级微胶囊破胶剂。纳米级微胶囊表面张力较低,能有效进入压裂缝中,使压裂液彻底破胶。Zhou等[10]采用原位界面共聚法在O/W型微乳液中制备了以过硫酸钾为核材、聚吡咯为壳材、粒径为400~750 nm的微胶囊。
然而,原位界面共聚法需要先配置分散相微乳液,再制备微胶囊破胶剂,制备方法复杂。笔者基于原位聚合法基本原理[11-13],直接将乳化剂、破胶剂与分散相混合,然后滴加作为壳材吡咯单体,通过原位聚合,制备得到粒径在100~300 nm的微胶囊破胶剂,并考察了其粒径分布、释放性能和破胶能力等性能。
1 实验部分
1.1 试剂与仪器
过硫酸铵、蒸馏水、无水乙醇、甘油、丙烯酰胺、乙酸、N,N-亚甲基双丙烯酰胺、亚硫酸氢钠,均为分析纯,国药集团化学试剂有限公司产品;司盘80、吐温60、吡咯、正丁醇,均为化学纯,国药集团化学试剂有限公司产品;5号白油,工业品,摩润克公司产品;蒸馏水、聚丙烯酰胺,实验室自制。
德国蔡司公司SUPER 55场发射扫描电子显微镜;德国布鲁克公司EQUINOX-55傅里叶红外光谱分析仪;湖南湘仪集团台式高速离心机H1850;上海雷磁有限公司DDS-307A电导率仪;美国博勒飞Brookfield DV-II旋转黏度计;英国Renishawn公司inVia Reflex显微共聚焦拉曼光谱仪;英国马尔文公司Nano-ZS90激光粒度仪。
1.2 微胶囊破胶剂及聚丙烯酰胺的制备
微胶囊破胶剂的制备:25 ℃下,将溶剂(40 mL 5号白油、0.24 g乙醇、0.16 g正丁醇)、乳化分散剂(2 g司盘80)混合均匀,搅拌条件下缓慢加入芯材4.0 g过硫酸铵。将上述混合物在300 r/min的条件下搅拌1 h后,加入乙酸,调节体系的pH值为4~5,滴加作为胶囊壳的材料(1.6 g吡咯)和润湿剂(1 mL的1%甘油水溶液)的混合物,反应4 h。反应结束后,静置沉淀,去除上层溶剂,得到黑色固体,用蒸馏水和无水乙醇清洗,真空50 ℃下干燥12 h,得到黑色固体即为含有甘油的过硫酸铵微胶囊,保存备用。
重复上述实验,改变润湿剂甘油水溶液添加量,分别添加0、2、3 mL质量分数为1%的甘油水溶液,制备不同甘油含量的过硫酸铵微胶囊。
聚丙烯酰胺的制备:将48 g白油、12 g司盘80、4 g吐温60混合均匀加到三口烧瓶中,通入氮气,加入0.5 g质量分数为10%的过硫酸铵、质量分数为0.1%的N,N-亚甲基双丙烯酰胺、24 g质量分数为60%的丙烯酰胺水溶液,搅拌10 min后,加入0.5 g质量分数为10%的亚硫酸氢钠引发剂,30 ℃水浴下反应1 h,得到半透明乳液。将乳液用乙醇沉淀并清洗数次后,在50 ℃真空干燥箱中烘干,得到白色粉末即为聚丙烯酰胺。配制质量分数为1%的聚丙烯酰胺水溶液备用。
1.3 过硫酸铵微胶囊的表征
使用德国蔡司SUPER 55场发射扫描电子显微镜对微胶囊进行表面形貌分析;使用傅里叶红外光谱分析仪与激光显微拉曼光谱仪对微胶囊进行结构表征。其中,拉曼光谱仪测试参数为:激光波长为532 nm,激光输出功率为500 mW,波数范围 100~3200 cm-1。
称取0.01 g微胶囊样品,离心10 min,干燥24 h后称重,干重与胶囊样品的比值即为微胶囊破胶剂的包裹率[13]。
在模拟地层温度(80 ℃)下,将不同甘油添加量的0.01 g微胶囊加入100 mL水中,使用电导率仪测定不同甘油含量的微胶囊样品在水中的电导率。
1.4 压裂液样品的性能评价
分别配制质量分数均为0.1%的微胶囊聚丙烯酰胺水溶液和过硫酸铵聚丙烯酰胺水溶液。以聚丙烯酰胺水溶液作为空白对比样品,室温下观察微胶囊在压裂液中的分布情况;通过激光粒度仪测量微胶囊破胶剂的粒径,观察其粒径分布。
取3组溶液样品,用旋转黏度计分别测量其在80 ℃下降解0、1、4、8 h时的黏度,绘制曲线并对比。并计算黏度保留率A[14]:
(1)
式中,μ1为溶液降解后黏度,mPa·s;μ0为溶液降解前黏度,mPa·s。
2 结果与讨论
2.1 过硫酸铵微胶囊结构性能分析
2.1.1 SEM分析
图1为微胶囊破胶剂样品的SEM图。由图1可观察到,微胶囊破胶剂呈球体,大小均匀,粒径在200 nm左右,微胶囊通过聚吡咯外壳彼此黏接。
2.1.2 红外与拉曼光谱分析
图2为微胶囊经过纯化处理后的红外光谱与拉曼光谱。由图2(a)可知:在1041 cm-1、615 cm-1处的峰对应过硫酸铵中的S=O和S-O键的特征吸收峰[10];1653 cm-1是聚吡咯中C=C键的振动吸收峰;1522 cm-1处的吸收峰为聚吡咯环中N-H的变形振动吸收峰;1457 cm-1处的峰为吡咯环中 C-N 的伸缩振动吸收峰[11];3200~3400 cm-1处的吸收峰为甘油中-OH伸缩振动吸收峰。由图2(b)可知:在1562 cm-1处为聚吡咯的C=C骨架特征吸收峰;1401 cm-1处的吸收峰为聚吡咯中C-N伸缩振动吸收峰[15];925 cm-1处为吡咯环中双极化子的变形振动吸收峰[16];810 cm-1为过硫酸铵中过氧键O-O键的特征吸收峰[17]。结合红外光谱与拉曼光谱分析结果,说明聚吡咯成功包覆了过硫酸铵。
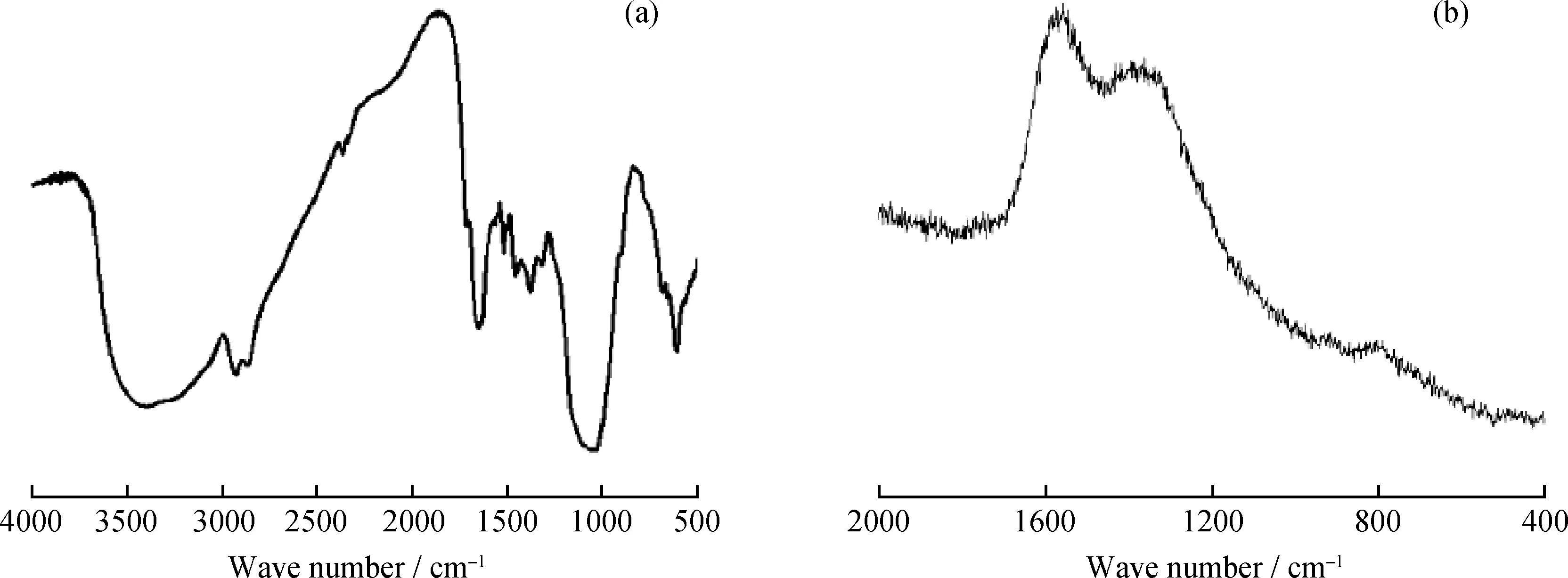
图2 过硫酸铵微胶囊红外光谱与拉曼光谱
2.1.3 不同甘油量的微胶囊包裹率
包裹率是评价原位聚合法能否经济高效制备过硫酸铵微胶囊的一个重要指标,直接反映聚吡咯是否成功包裹了过硫酸铵。微胶囊包裹率测定结果表明,分别添加0、1、2、3 mL质量分数为1%的甘油水溶液制备微胶囊对应的包裹率分别为96%、93%、93%、88%。这说明,随着甘油水溶液添加量的增加,微胶囊的包裹率逐渐降低,原因在于微胶囊形成的过程中甘油参与了微胶囊壳的形成。在微胶囊形成过程中,聚吡咯在固体过硫酸铵颗粒表面形成聚合物膜;该膜有很多微孔隙,具有一定的孔隙度和渗透率;吡咯聚合形成胶囊壳过程中,作为润湿吸水剂的甘油吸附在聚吡咯膜的孔隙中,使得其部分孔隙亲水。当微胶囊溶于水中时,在毛管力作用下,胶囊壳孔隙中的甘油吸水溶解,过硫酸铵更易从孔隙释放,而没有甘油吸附的微孔隙,水较难进入,微胶囊释放慢。因此,甘油加量多时,胶囊壳上亲水孔隙多、吸水快,过硫酸铵释放也快,因此造成微胶囊的包裹率降低。
文献[5]以氯仿为溶剂、以十二烷基硫酸钠/聚乙二醇6000为乳化剂,制备的过硫酸铵微胶囊最高包裹率为91.80%。与之相比,以5号白油为溶剂,司盘80为乳化分散剂原位聚合微胶囊制备方法得到的微胶囊包裹率较高,说明笔者制备微胶囊的方法是可行的。
2.1.4 过硫酸铵微胶囊水分散液的电导率
过硫酸铵微胶囊水分散液的电导率表征微胶囊的释放情况。由于壳材聚吡咯的亲水性差,甘油亲水性好,所以在聚吡咯中掺杂甘油水溶液来控制过硫酸铵的释放速率[10]。图3为不同甘油水溶液添加量的微胶囊水分散液在80 ℃时的电导率随时间变化曲线。其中,曲线(1)是过硫酸铵在水中的释放曲线;(2)、(3)、(4)、(5)分别对应添加3、2、1 mL质量分数为1%的甘油及无甘油添加的微胶囊在水中释放的电导率曲线。由图3中曲线(1)可知,随着时间的延长,过硫酸铵在水中的电导率不断增加,其中,在0~3 h内过硫酸铵水溶液的电导率增加较快,原因在于过硫酸铵在80 ℃水中分解的半衰期为2.1 h[18],其受热分解产生2个自由基和1个不反应的硫酸根离子[19],使其电导率在0~3 h迅速增加。在3~8 h内过硫酸铵分解减缓,导致电导率增加缓慢。
由图3中曲线(2)~(5)可知,在80 ℃水中,随着释放时间的增加,微胶囊的电导率逐渐提高,表明微胶囊能缓慢释放过硫酸铵,且微胶囊的电导率随着甘油添加量的增加而增大。对比曲线(2)、(5),未添加甘油时(曲线(5)),微胶囊溶液电导率在 0~8 h 内增长缓慢,说明微胶囊释放缓慢;添加甘油后在0~1 h内,胶囊溶液的电导率明显增大,说明甘油的添加明显加快了微胶囊的释放。但对比过硫酸铵在水中释放的电导率(曲线(1)),微胶囊释放过硫酸铵是一个缓慢过程,微胶囊制备成功。比较曲线(2)、(3)、(4)可知,随着添加甘油的量增加,电导率升高加快,说明微胶囊的吸水速率提高,释放过硫酸铵的速率也越快。
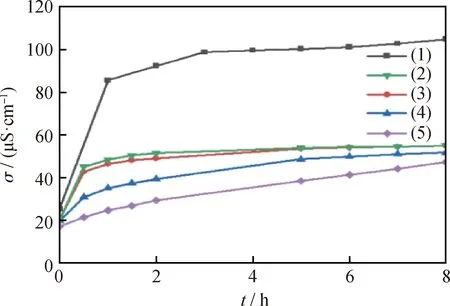
图3 80 ℃时不同甘油添加量的微胶囊水分散液的电导率随时间变化曲线
2.2 过硫酸铵微胶囊的形成过程及在水中的释放过程
过硫酸铵微胶囊的形成与乳化剂种类有关,司盘80是一种适宜的乳化分散剂。其溶于白油溶剂中,亲水基团作用于过硫酸铵[10],疏水基团作用于吡咯单体,使吡咯单体在过硫酸铵表面积聚。吡咯单体的氧化电势较低,过硫酸铵迅速引发吡咯单体发生聚合反应生成聚吡咯为壳、过硫酸铵为芯的过硫酸铵微胶囊。
根据微胶囊壳材渗透性的不同,过硫酸铵微胶囊的释放方式主要有2种:应力挤压破碎释放和渗透释放。当微胶囊采用防水、防渗的惰性材料为壳时,水分无法进入;在压裂操作后,压裂缝闭合产生闭合应力,胶囊壳在应力挤压作用下发生形变破裂而释放内部破胶剂,因此其释放为应力挤压释放。当破胶剂外壳为可渗透性材质时,水通过微胶囊上的孔隙缓慢进入微胶囊,破胶剂溶解形成水溶液,通过扩散缓慢渗透到压裂液中进行破胶[20]。由于笔者制备的微胶囊壳中含有的甘油具有吸水性,使微胶囊壳具有可渗透性,因此其释放过程为渗透释放。
图4为过硫酸铵微胶囊在水中的释放过程示意图。由图4可知:图4(a)为微胶囊的初始状态,胶囊壳中存在微孔隙,在干燥情况下,微胶囊是稳定的,不会释放;当微胶囊处于水环境中,水通过聚吡咯外壳中掺杂的甘油进入微胶囊,导致微胶囊吸水膨胀,固体过硫酸铵遇水溶解(图4(b));溶解后的过硫酸铵通过浓差扩散从微孔隙释放,进入溶液本体,见图4(c)。结合2.1.3节微胶囊包裹率和2.1.4节微胶囊溶液电导率分析可知,甘油水溶液添加量过多,包覆效果不好,包裹率较低;甘油水溶液添加量过少,微胶囊释放速率低。因此,添加2 mL质量分数为1%的甘油水溶液制得的微胶囊具有较高的包裹率和较好的释放效果。
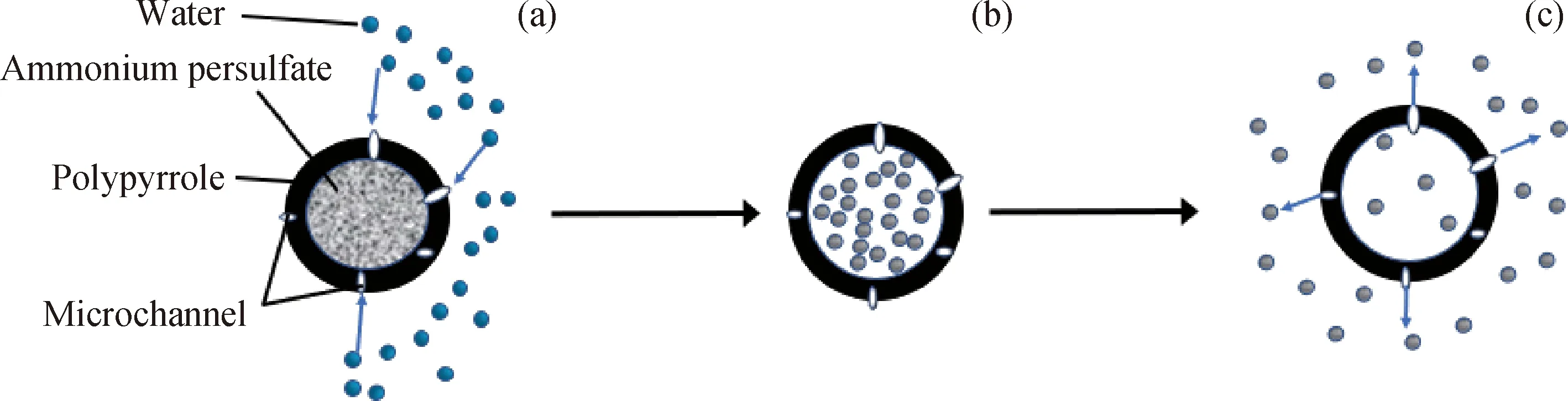
图4 微胶囊在水中释放过程示意图
2.3 微胶囊破胶剂的应用性能
2.3.1 微胶囊在压裂液中的分散性能
压裂液由微胶囊和聚合物水溶液组成。实验以添加2 mL甘油水溶液制得的微胶囊为对象考察微胶囊在压裂液中的分散性能,图5为质量分数为0.1%微胶囊悬浮分散在质量分数为1%聚丙烯酰胺水溶液中微胶囊的分散情况及其粒径分布。由图5(a)可知,微胶囊悬浮液分散均匀,在室温下放置 7 d 不分层。由图5(b)可知,微胶囊粒径集中分布在100~300 nm,主要在200 nm左右,与图1结果一致,说明微胶囊在压裂液中的分散性能良好;但仍有少量微胶囊的粒径在400 nm左右。这是因为在微胶囊制备过程中过硫酸铵颗粒出现聚集和沉降[21],聚集的过硫酸铵粉末粒度较大,则制备的微胶囊粒径也较大;同时,在过硫酸铵引发吡咯聚合反应过程中,会出现少量多核微胶囊,其粒径也相对较大。
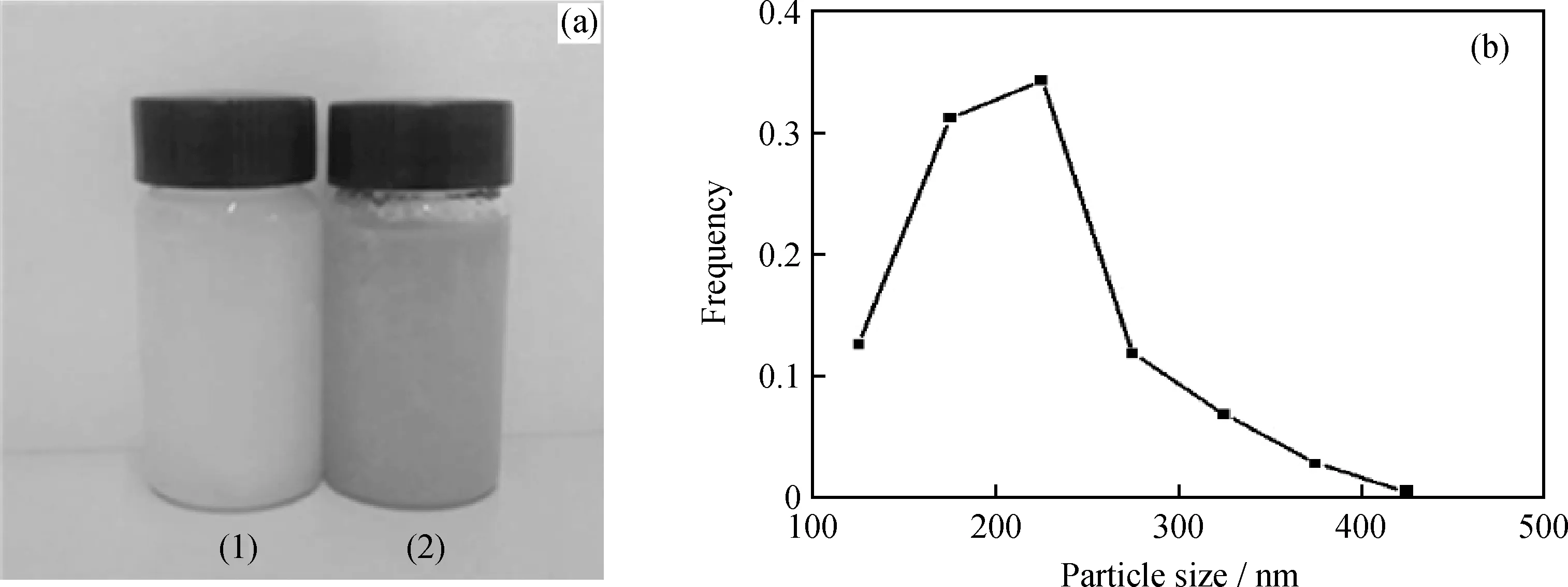
图5 微胶囊在聚合物中的悬浮分散图及其在水中的粒径分布
2.3.2 压裂液的延迟破胶性能
聚丙烯酰胺是一种网状结构微凝胶。压裂液破胶,即破坏其网状凝胶结构,降低黏度利于返排。压裂液是否破胶,一般依据其黏度变化来判断[22]。图6为添加质量分数0.1%的微胶囊聚合物溶液、过硫酸铵聚合物溶液和空白聚合物溶液在模拟地层温度(80 ℃)、不同时间下,黏度随剪切率的变化曲线。由图6(a)可知:微胶囊聚合物溶液、空白聚合物溶液的黏度较高,但随着剪切率的增加而逐渐降低;而过硫酸铵聚合物溶液的黏度较低,但随剪切率的变化降低缓慢。原因在于随剪切率增加,微胶囊的聚合物溶液和空白聚合物溶液中聚合物的三维网络结构被破坏,黏度降低;过硫酸铵的聚丙烯酰胺黏度较低,是因为过硫酸铵分解成了高反应活性的硫酸根离子,引起自由基氧化断链反应,使大分子聚丙烯酰胺降解为小分子链,而剪切作用对小分子链的影响较小。微胶囊的聚合物溶液黏度较空白聚合物溶液低,说明微胶囊在聚丙烯酰胺溶液中存在缓慢释放过程。
对比图6(a)、(d)中空白聚合物的黏度可知,聚合物在模拟地层温度下会自发降黏。这是由于聚合物溶液中存在少量溶解氧,聚合物发生热解氧化反应链断裂,黏度降低[23]。但是黏度降低速率慢,降解8 h后黏度只能达到20~30 mPa·s,达不到《压裂液通用技术条件》(SY 6376—2008)中规定的返排标准(5 mPa·s以下)。在聚合物中加入过硫酸铵,聚合物黏度降低过快,1 h后破胶液就达到返排标准。
在聚合物溶液中加入等量微胶囊,在0、1、4、8 h时黏度分别如图6(a)~(d)所示:前期(0、1 h)与空白聚合物溶液相差较小;中期(4 h)与聚合物黏度相近(200 mPa·s),满足现场压裂施工的要求[9];在8 h时黏度降到5 mPa·s以下,满足返排标准。空白聚合物压裂液前期降解速率较快,后期降解较慢,黏度降至5 mPa·s以下需要24 h[24],超过《压裂液通用技术条件》(SY 6376—2008)中压裂液最长破胶时间12 h。压裂过程中时间越长,压裂液侵入地层越多越深,对地层伤害越大,因此控制压裂作业的时间对成功压裂具有重要意义。加入微胶囊的意义在于能够根据压裂施工设计时间的变化,加入不同释放速率的微胶囊以调节压裂液的破胶时间。由以上结果可以看出,制备的过硫酸铵微胶囊具有延迟降黏能力,能使压裂液延缓破胶7 h以上。
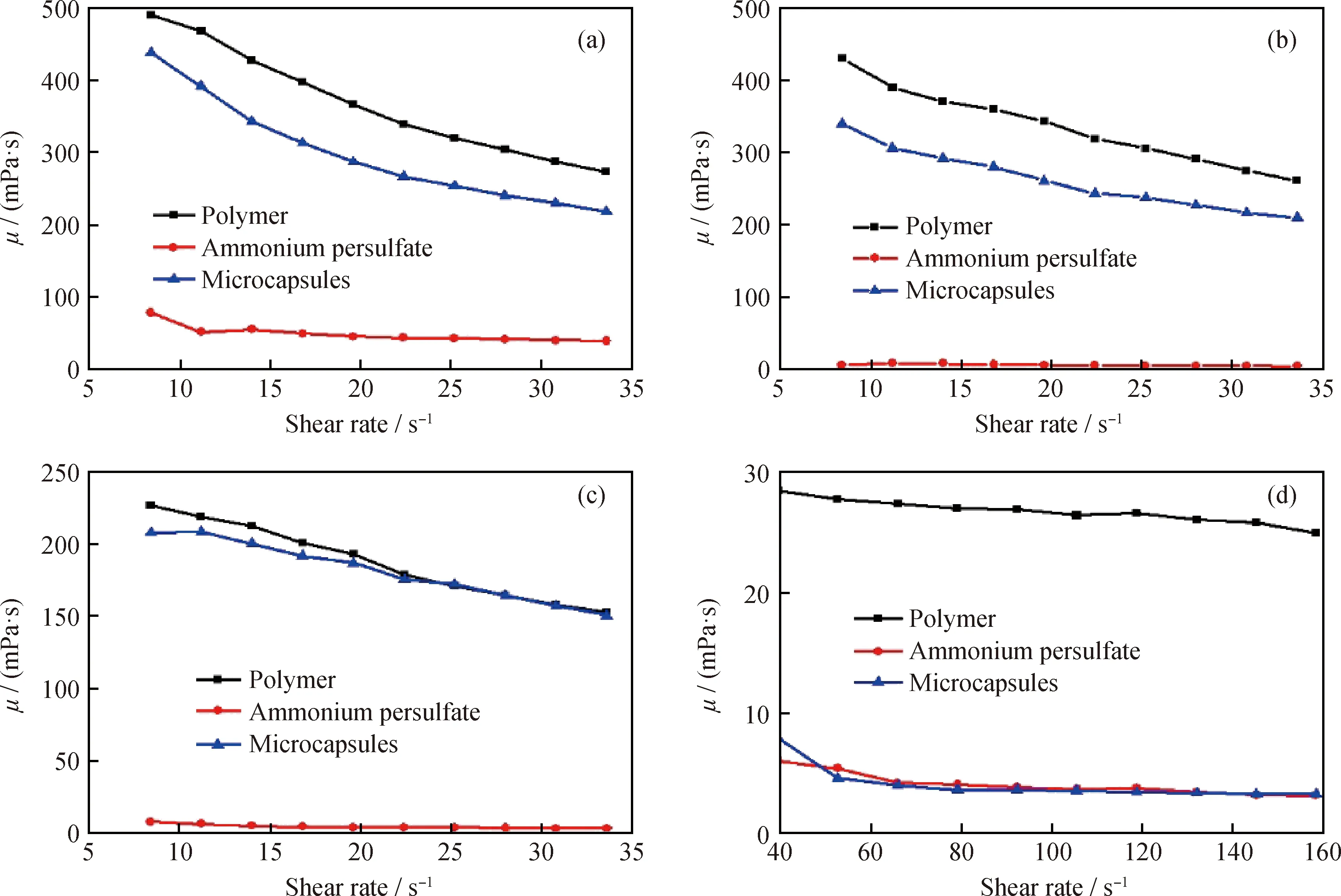
图6 不同时间3种样品黏度随剪切率的变化
图7为3种样品黏度保留率和时间的关系。由图7可知:空白聚合物在80 ℃下有较好的黏度稳定性,降解1 h黏度保留率大于90%。加入质量分数0.1%过硫酸铵后,样品破胶很快,黏度保留率仅为13.2%,1 h后保留率降至2%。加入等量微胶囊后,样品黏度初期保留率较高;降解4 h后保留率达49.4%,与聚合物降解的黏度保留率相近;降解8 h后黏度保留率降至2.6%,压裂液完成破胶。这说明用过硫酸铵微胶囊作为破胶剂能使压裂液在施工过程中保持较高的黏度,有利于前期压裂,又能在压裂施工结束后使压裂液快速破胶返排。
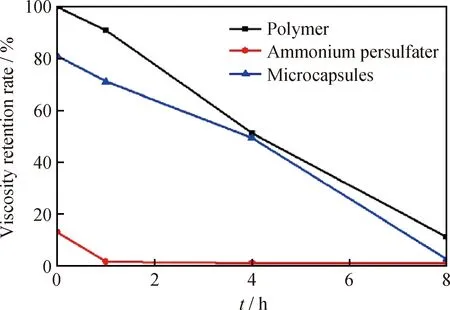
图7 3种样品在不同降解时间黏度保留率曲线对比
3 结 论
(1)以5号白油为分散介质,司盘80为乳化剂,以过硫酸铵为芯材、聚吡咯为壳材,通过原位聚合法成功制备了平均包裹率为92.5%的微胶囊。
(2)电导率实验表明,微胶囊的释放过程为渗透释放,且随着甘油量的增多,聚吡咯外壳中的微孔隙中甘油增多,形成亲水孔隙越多,吸水速率越大,固体过硫酸铵溶解越快,释放速率越大。
(3)通过高温降解对照实验表明,微胶囊可以使聚合物溶液在降解4 h内保持较高的黏度,且降解 8 h 后黏度达到返排标准。该过硫酸铵微胶囊可以使压裂液延缓破胶7 h以上。
