基于可逆离子氢键构筑室温快速自修复聚硅氧烷弹性体
2020-09-25时金凤符文鑫李志波
时金凤, 赵 娜, 符文鑫, 李志波
(1.青岛科技大学高分子科学与工程学院,山东 青岛 266042;2.中国科学院化学研究所先进高分子材料实验室,北京 100190)
聚硅氧烷弹性体是一类性能优异的材料,其生物相容性好,力学性能易调节,耐化学腐蚀性优异,在较宽的温度范围内拥有良好的弹性,被广泛应用于柔性电子器件[1]、耐高低温密封材料[2]、人造血管[3]等方面。对比传统的热固性弹性体材料,自修复材料具有修复损伤的性能,从而延长了产品的使用寿命,某种程度上能够减少材料的浪费[4]。
近年来,许多文献报道了赋予材料自修复功能的不同方法,从而拓展了其在涂料[5]、黏合剂[6]、传感器[7]、柔性电子器件[8]等领域的应用。制备自修复型聚硅氧烷弹性体的方法主要有:(1)植入微胶囊[9],首先制备能够包覆催化剂的微胶囊,再将微胶囊埋入材料基体。当材料受外力作用发生破坏时,应力诱导微胶囊破裂,催化剂从胶囊中释放,分散在材料基体中的预聚体在催化剂作用下发生化学反应,进而实现断裂处自修复。但此时微胶囊的尺寸和分散性等会影响弹性体最终的力学性能,且不能实现多次自修复。(2)引入可逆共价键,如在聚合物基体中引入亚胺键[10]、双硫键[11]、Diels-Alder 反应形成可逆的化学键[12,13],以及硼酸酯或硼氧六环键[14,15]等。可逆共价键在保证材料力学性能的基础上赋予材料自愈合性能,同时也避免了植入微胶囊引入的化学污染。但实现可逆共价键通常需要外界条件的充分刺激,比如长时间或深层次的光、热刺激等,并且需要相对复杂的单体分子结构设计和合成。(3)引入非共价键,如氢键[16]、离子键[17]、金属配位键[18-20]等。与前两种方式相比,非共价键的键能较弱,易于断裂与重组,当材料出现断裂缺口时,更易通过非共价键的重组实现自修复。另外,利用可逆共价键和非共价键或多种类型非共价键的协同作用,可以同时提高材料的力学强度和可拉伸性能。例如,Zhang 等[21]报道了含有脲基和双硫键交联的聚硅氧烷弹性体,体系中大量的脲键形成分子链内/间的氢键弱相互作用,当材料发生大形变时进行能量耗散,从而赋予材料优异的柔韧性;另外,可逆的双硫键可维持聚合物网络的完整性,提高材料的力学强度。Kang 等[22]将6-苯基-2,2’-二吡啶官能团引入聚硅氧烷主链,利用环二价铂金属络合物之间Pt-Pt 和π-π 两种非共价相互作用形成交联网络,制备的弹性体表现出优异的柔韧性;但是多种化学键的引入也增加了材料设计、合成以及性能调控的难度。
通常,未补强的聚硅氧烷弹性体的断裂伸长率小于300%[23],而且这类材料是基于硅氢加成反应实现共价交联的聚硅氧烷弹性体,并不可回收和自修复。因此,探索简单高效的方法制备高度可拉伸、可回收、可快速自修复的聚硅氧烷弹性体材料对于拓宽聚硅氧烷材料在下一代智能材料领域(如高分子驱动器、软体机器人、电子皮肤等)的应用具有重要意义。
基于上述研究背景,本文通过实验室自制的环三聚磷腈碱(CTPB)[24,25]作为催化剂,在温和条件下高效合成了具有不同分子量和硅氧乙烯基单元摩尔分数(χ,连接有C=C 双键的Si―O 单元占全部Si―O 单元的比例)的线型聚硅氧烷前驱体,并通过硫醇-烯点击化学反应,简单高效地制备了侧基分别为羧基和氨基的聚硅氧烷(PDMS-g-COOH 和PDMS-g-NH2)。将两种聚合物溶液充分均匀混合,成功制备了由羧基与氨基间离子氢键交联的聚硅氧烷弹性体(PDMS-g-[COOH/NH2]),进而研究了聚合物基体中乙烯基含量、聚合物分子量对弹性体拉伸性能、自修复性能的影响。结果表明,PDMS-g-[COOH/NH2]弹性体具有良好的伸展性能,且在室温条件下可以实现快速自修复。本研究为制备可低能量驱动自修复的聚硅氧烷弹性体提供了一种简单通用的制备方法。
1 实验部分
1.1 原料和试剂
八甲基环四硅氧烷(D4)、四甲基四乙烯基环四硅氧烷(V4):纯度98.0%,上海阿拉丁生化科技股份有限公司,使用前加入氢化钙常温下搅拌24 h,减压蒸馏,并收集在盛有分子筛的存储瓶密封保存;封端剂四甲基二乙烯基硅氧烷(纯度97%)、巯基丙酸(纯度99%):上海阿拉丁生化科技股份有限公司;CTPB:实验室自制;巯基乙胺盐酸盐(纯度98%)、光引发剂2,2-二甲氧基-苯基苯乙酮(DMPA,纯度99%):安耐吉有限公司;甲苯:分析纯,烟台远东精细化工有限公司,使用前经有机纯化系统纯化除水;四氢呋喃、甲醇:分析纯,天津富宇精细化工有限公司。
1.2 测试与表征
凝胶渗透色谱(GPC)仪(美国,Agilent 1260):以单分散聚苯乙烯(PS)为标样,测试温度为40 ℃,THF 为流动相,流速为1.0 mL/min;红外光谱(FT-IR)仪(德国,Bruker Tensor 27):测试波长范围600~4 000 cm-1,采用衰减全反射(ATR)模式;核磁共振波谱(NMR)仪(瑞士,Bruker AVANCE NEO 400 MHz):溶剂为氘代氯仿(CDCl3)、氘代甲醇(CD3OD)、氚代水(HOD);拉力试验机(美国,Instron 5900):拉伸速率分别为50 、100 、200 mm/min,试验样条长为4 cm,宽为3 mm。
1.3 实验步骤
1.3.1 甲基乙烯基线型聚硅氧烷(PDMS-g-Vi)的合成 首先,称取24 mg(0.02 mmol)CTPB 置于单口瓶中并加入1 mL 甲苯充分搅拌溶解,在另外一个史莱克瓶加入计量好的D4、V4、封端剂(具体配比如表1 所示)和适量甲苯充分溶解。然后,将CTPB 的甲苯溶液转入上述单体混合溶液中,30 ℃下搅拌反应30 min 后,将反应液缓慢倒入50 mL 甲醇中沉降析出聚合物,用甲醇充分洗涤3 次,去除小分子和寡聚物。最后,于真空烘箱中室温真空干燥得到目标聚合物PDMS-g-Vi,其合成路线如图1(a)所示。根据调整投料比(n(D4)/n(V4))与聚合物在体系中的浓度(如表1 所示),得到了3 种线型聚硅氧烷前驱体(P1,P2,P3)。

表1 CTPB 催化D4 和V4 开环共聚合结果Table 1 Ring-opening polymerization of D4 and V4 catalyzed by CTPB
1.3.2 PDMS-g-COOH 的合成 以P1-g-COOH 为例:称取2.5 g P1(5 mmol 乙烯基)和50 mL THF 置于100 mL史莱克瓶中,氮气鼓泡排除体系中的空气。在氮气保护下,将25 mg(0.1 mmol)光引发剂DMPA 和1.6 g(15 mmol)巯基丙酸加入到上述溶液中混合均匀(巯基丙酸与乙烯基的摩尔比为3),室温条件下,汞灯光照30 min 后,将聚合物的THF 溶液倒入去离子水中沉降,重复水洗3 遍,除去多余的巯基丙酸,室温下真空干燥得到P1-g-COOH。通过1H-NMR 确定接枝率大于99%,其合成过程如图1(b)所示。
1.3.3 P1-g-NH2的合成 称取2.5 g P1(5 mmol 乙烯基)和30 mL THF 置于100 mL 史莱克瓶中,氮气鼓泡排除体系中的空气。在氮气保护下,将25 mg(0.1 mmol)光引发剂DMPA、1.7 g(15 mmol)巯基乙胺盐酸盐的甲醇(6 mL)溶液加入到上述溶液中混合均匀(巯基丙酸与乙烯基的物质的量之比为3),室温条件下,汞灯光照30 min 后,聚合物的THF 溶液用饱和碳酸钠水溶液中和,倒入去离子水中沉降,重复水洗3 次,除去多余的巯基乙胺盐酸盐,室温下真空干燥得产物P1-g-NH2。通过1H-NMR 确定接枝率大于99%,其合成过程如图1(b)所示。
1.3.4 PDMS-g-[COOH/NH2]弹性体的制备 以P1-g-[COOH/NH2]弹性体为例:将等物质的量的P1-g-COOH与P1-g-NH2分别溶于THF 中,质量浓度约为10 mg/mL。将P1-g-COOH 的THF 溶液逐滴加入到PDMS-g-NH2的THF 溶液中,搅拌过夜。将得到的黏稠聚合物溶液倒入四氟乙烯模具中,室温过夜,溶剂自然挥发后,放入真空干燥箱进一步除去溶剂,得到P1-g-[COOH/NH2]弹性体。采用相同的方法将P2-g-COOH、P3-g-COOH 分别与P1-g-NH2制备得到另外两种聚合物弹性体,命名为P2-g-[COOH/NH2]弹性体和P3-g-[COOH/NH2]弹性体。样品的制备过程如图1(c)所示。
2 结果与讨论
2.1 PDMS-g-Vi 的表征
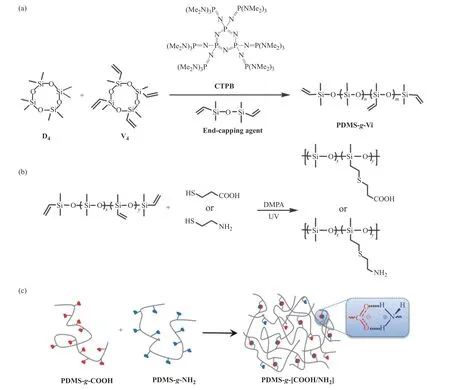
图1 (a)PDMS-g-Vi,(b)P1-g-COOH 与P1-g-NH2,(c)PDMS-g-[COOH/NH2]的合成示意图Fig.1 Synthetic routes of (a)PDMS-g-Vi,(b)P1-g-COOH and P1-g-NH2,(c)PDMS-g-[COOH/NH2]
PDMS-g-Vi 的数均分子量(Mn)、分子量分布(Đ)通过GPC 表征,χ 通过核磁共振氢谱表征,结果如表1所示。一般由碱性阴离子引发的环硅氧烷开环聚合最终会达到平衡,得到的聚合物中混有大量的线型寡聚物和不同分子量的环硅氧烷中间体,导致聚合物的分子量分布较宽(Đ>2)[26,27]。然而用CTPB 催化开环聚合的结果显示出了高单体转化率(>80%)和相对较窄的分子量分布(Đ<2),符合非平衡阴离子开环聚合的特点[28]。此外,当投料比n(D4)/n(V4)=85/15 时,通过调整聚合物在体系中的浓度,由1.5 mol/L 增加到2.5 mol/L,可以成功制备乙烯基摩尔分数相似,数均分子量分别为43.0×103和144.8×103的P1 和P2。通过调节n(D4)/n(V4)由85/15 增加至92/8,保持聚合物在体系中的浓度为2.5 mol/L,制备的P2 和P3 其Mn相近,但χ 分别为14.5%和7.1%。
2.2 改性聚硅氧烷及其弹性体的结构表征
图2 是P1、P1-g-COOH 和P1-g-NH2的1H-NMR 谱图。在聚硅氧烷前驱体P1 的1H-NMR 谱图中,化学位移5.70~6.05(b, c)处的多重峰归属于乙烯侧基(CH2=CH―)的氢原子信号。经巯基丙酸改性以后的P1-g―COOH,在0.85(e)、2.47~2.70(g, f)和2.70~2.84(h)处出现的新峰分别归属于3种亚甲基(―Si―CH2―,―CH2―S―CH2―,COOH―CH2―)上氢的化学位移;同时,乙烯侧基在5.70~6.05(b, c)处的峰也完全消失,说明了P1-g-COOH 的成功合成。P1-g-NH2的结果与P1-g-COOH 的结果类似,与P1 对比,0.85(i)、2.49~2.66(k, j)、2.78~2.92(l)处出现的新信号峰分别归属于―Si―CH2―、―CH2―S―CH2―、NH2―CH2―的化学位移,并且乙烯侧基的信号峰完全消失,也证明了硫醇-烯加成反应的完成。另外,P2、P3及羧基改性的产物P2-g-COOH 和P3-g-COOH 也显示出类似的结果。
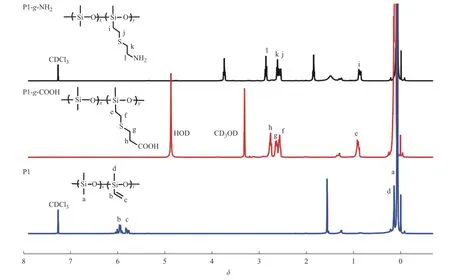
图2 样品的1H-NMR 谱图Fig.2 1H-NMR spectra of samples
P1、 P1-g-COOH 和 P1-g-NH2的 FT-IR 结 果(图(3))也证实了硫醇-烯加成反应的成功。如图3所示,前驱体P1 在1 598 cm-1和3 054 cm-1处分别显示出乙烯侧基C=C 和C―H 的伸缩振动特征吸收峰[29,30]。经过巯基丙酸(红线)改性后,P1-g-COOH在1 714 cm-1处出现新的吸收峰,归属于引入的羧基官能团C=O 的伸缩振动峰[29],并且在1 598 cm-1和3 054 cm-1处C=C 双键上的特征吸收峰消失,进一步说明了P1-g-COOH 的成功合成,与图2 结果吻合。同时,巯基乙胺(蓝线)改性后的P1-g-NH2的FT-IR 谱图于1 588 cm-1处显示宽吸收峰,归属于引入氨基的N―H 弯曲振动[31],也证明了P1-g-NH2成功合成。此外,交联弹性体P1-g-[COOH/NH2]的FTIR 结果显示C=O 在1 714 cm-1处伸缩振动吸收峰右移,与对应N―H 在1 572 cm-1处的弯曲振动峰形成新的宽吸收峰,充分说明COOH 与NH2之间存在相互作用形成了离子氢键。聚合物P2 和P3、羧基改性产物P2-g-COOH 和P3-g-COOH,以及其与P1-g-NH2形成的弹性体的FT-IR 结果也显示出类似的结果。
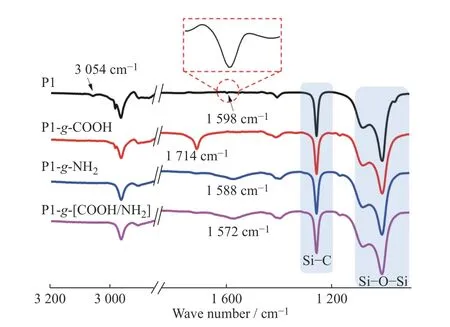
图3 样品的FT-IR 谱图Fig.3 FT-IR spectra of samples
2.3 PDMS-g-[COOH/NH2]弹性体的力学性能
当体系中存在大量动态结合的离子氢键时,氢键可为材料提供可观的力学性能和优异的延展性。作为一种中等或强氢键,离子氢键的键能为5~35 kJ/mol,仍远低于共价键和离子键,属于非共价弱键相互作用[32]。PDMS-g-[COOH/NH2]弹性体的交联网络正是基于羧基和氨基的离子氢键,如图1(c)所示。
图4 为P1-g-[COOH/NH2]弹性体在不同拉伸速率下的应力-应变曲线。由图可知,P1-g-[COOH/NH2]弹性体的力学性能对拉伸速率有很强的依赖性。随着拉伸速率的增加,弹性体的最大伸长率降低。在低拉伸速率(50 mm/min)条件下,P1-g-[COOH/NH2]弹性体中聚硅氧烷主链可以缓慢松弛,同时分子链间离子氢键的断裂速率与重组速率相匹配,即断裂的离子氢键具有足够的时间完成重组,弹性体的交联网络仍旧得以保持,表现出较好的延伸性,其断裂伸长率可达到877.0%。当拉伸速率依次增大到100 mm/min 和200 mm/min 时,其对应的最大断裂伸长率分别降低到733.4%和522.3%。这是因为分子链在短时间内产生很大滑移,由拉伸应力所产生的部分断裂的离子氢键难以实现重组,致使样品断裂。另外,根据时温等效原理,随拉伸速率的增加,分子链达到相应应变所需的时间缩短,短时间尺度高分子网络的力学松弛行为表现出类似于热固性材料的弹性行为,所以拉伸断裂强度也逐渐增加,断裂伸长率也随之降低。
图5 为PDMS-g-COOH 与P1-g-NH2所形成的弹性体在50 mm/min 拉伸速率下的应力-应变曲线。结果显示:在相同的拉伸速率下,通过改变聚合物的分子量、离子氢键的交联密度,可以调整所得弹性体的拉伸性能。在χ 接近的情况下(表1 中的P1 和P2),相应弹性体的拉伸强度从230.9 kPa 增加到344.5 kPa,同时断裂伸长率也从877.0%增加到1 038.1%。聚硅氧烷链段分子量增加,分子链间的缠结程度增大,从而在一定程度上限制了分子链的滑移,提高了材料的力学强度;同时,在交联密度相近的条件下,分子量越大的聚合物,其延展性能越好。另外,当聚合物数均分子量相近时,随着χ 从P3 的7.1%增加到P2 的14.5%,相应弹性体的拉伸强度由231.9 kPa 提高到344.5 kPa,说明增加聚合物分子链间的离子氢键交联密度能够增强弹性体网络的刚性,提高聚合物的拉伸强度,但柔韧性下降,断裂伸长率由1 394.7%降低到1 038.1%。
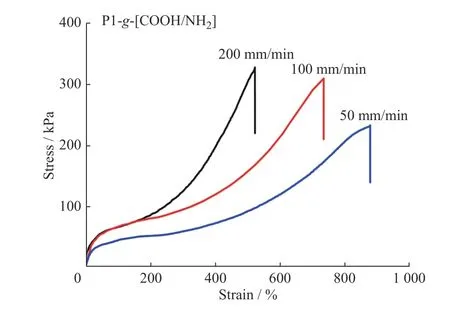
图4 P1-g-[COOH/NH2]弹性体在不同拉伸速率下的应力-应变曲线Fig.4 Tensile stress-strain curves of P1-g-[COOH/NH2] elastomer at different stretching speeds
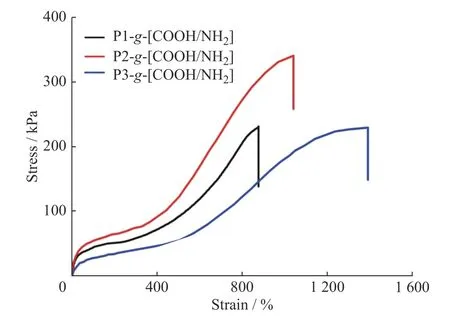
图5 PDMS-g-[COOH/NH2]弹性体在 拉伸速 率50 mm/min条件下的应力-应变曲线Fig.5 Tensile stress-strain curves of PDMS-g-[COOH/NH2]elastomers at the stretching speed of 50 mm/min
2.4 PDMS-g-[COOH/NH2]弹性体的自修复性能
文献中报道基于可逆共价键的自愈合弹性体大多需要外界辅助条件(如:高温[21]、UV[10]等)才可以进行自修复,且修复效率依赖于外界能量输入。因此,对低能量驱动的自修复材料的研究具有重要应用价值。基于离子氢键的弱相互作用,其相对更容易的动态成键/断键为构筑可室温快速自修复材料提供了一种可能。以P1-g-[COOH/NH2]弹性体为例,本文对其在室温下的自修复性能进行了研究。如图6(a)所示,首先将样品裁成两段,将两段样条在切口处对齐并紧贴在一起后,在室温下分别静置15 min 和30 min,无其他外部刺激,然后测其拉伸性能,结果如图6(b)所示。
将修复后样条的断裂强度与原始样品断裂强度的比值定义为弹性体的修复效率。从应力-应变结果可以看出,P1-g-[COOH/NH2]弹性体的自修复过程非常快。静置15 min 后,修复效率可以达到79%;静置30 min后,达到99%,且拉伸强度完全恢复到原始样条的拉伸强度,说明P1-g-[COOH/NH2]弹性体的自修复效率随时间的增加而增加。P1-g-[COOH/NH2]弹性体的快速自修复性能归因于构成网络结构的COO-/NH3+离子氢键在受到外力时动态可逆的快速成键,同时也得益于主链Si―O―Si 键良好的柔顺性,使得侧基官能团有较好的结合效率。当把P1-g-[COOH/NH2]弹性体的断裂面处重新组合在一起时,游离态的COO-与NH3+基团能够非常迅速地重组,形成新的离子氢键,从而实现材料的自修复。
3 结 论
(1)利用CTPB 催化D4与V4开环共聚制备不同数均分子量和不同硅氧乙烯基单元摩尔分数的线型聚硅氧烷,进而通过硫醇-烯点击化学反应,简单高效地成功合成侧基羧基或氨基改性的聚硅氧烷PDMS-g-COOH、PDMS-g-NH2,最后利用侧链上羧基与氨基之间的离子氢键交联形成自修复弹性体PDMS-g-[COOH/NH2] 。
(2)PDMS-g-[COOH/NH2]弹性体的力学强度和拉伸性能可通过改变聚合物的分子量、离子氢键交联密度来调节。
(3)此类离子氢键交联的聚硅氧烷弹性体在室温下具有快速自修复性。断裂样品在室温下静置愈合30 min后,其拉伸强度和断裂伸长率可完全恢复到原始样品的相应水平。
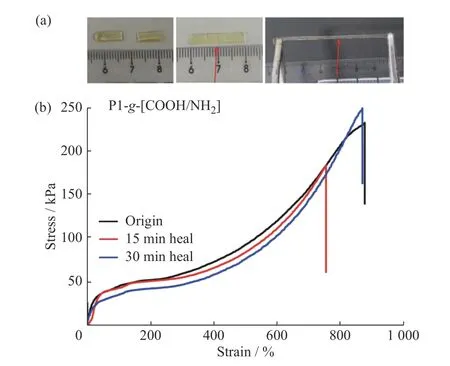
图6 (a)P1-g-[COOH/NH2]弹性体室温自修复过程;(b)样品修复前后的应力-应变曲线(拉伸速率为50 mm/min)Fig.6 (a)Self-healing behavior of P1-g-[COOH/NH2] elastomer at room temperature;(b)Tensile stress-strain curves of P1-g-[COOH/NH2] elastomer before and after healing(the stretching speed was 50 mm/min)
