高效液相色谱法测定硼替佐米中异戊醛含量
2020-09-24张德广王启帅
张德广,李 贞,刘 霞,王启帅
(1. 扬子江药业集团上海海尼药业有限公司,上海 浦东 201318;2. 扬子江药业集团,江苏 泰州 225321)
多发性骨髓瘤主要是因为骨髓浆细胞异常克隆增殖所致的一种恶性浆细胞病,在造血系统肿瘤中发病率占比约为 13%,且具有临床治疗效果差,治疗后复发率高等特点[1]。已有多项研究表明:在临床治疗多发性骨髓瘤中,应用硼替佐米药物可以取得良好治疗效果[2-3]。原料药的质量与制剂的质量息息相关,硼替佐米原料药在存储过程中会降解产生异戊醛(图 1),而异戊醛作为一种潜在基因毒性杂质,会对原料药的质量产生影响[4-5]。因此,建立硼替佐米中异戊醛的分析方法对于原料药的质量控制十分必要。根据 ICH M7《评估和控制药物中 DNA 反应性(致突变)杂质以限制潜在致癌风险》等相关指导原则及注射用硼替佐米的用法用量,得出硼替佐米原料药中异戊醛的限度为质量分数 0.03%。
异戊醛无明显紫外吸收,无法用紫外检测器直接测定,同时由于硼替佐米对热不稳定,会降解产生异戊醛,不宜采用顶空-气相法测定。2, 4-二硝基苯肼作为一种常用的检测羰基化合物(-CO)的试剂,在酸性介质中,羰基化合物与 2, 4-二硝基苯肼反应,生成具有紫外吸收的 2, 4-二硝基苯腙,从而可以采用液相色谱检测,间接测定样品中的醛类。本研究利用上述原理,检测硼替佐米中的异戊醛。
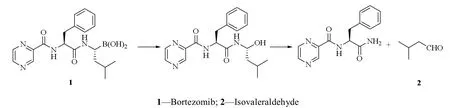
Fig. 1 Degradation pathways of bortezomib图 1 硼替佐米降解途径
1 仪器与材料
1260 高效液相色谱仪(配四元梯度泵,DAD 检测器和 Empower®3 色谱工作站,美国,安捷伦公司),XP205 电子分析天平(瑞士,梅特勒-托利多集团)。
异戊醛对照品(质量分数为 99.78%,批号 20151227,天津市光复精细化工研究所),硼替佐米原料药(批号 15012801,扬子江药业集团江苏海慈生物医药有限公司),乙腈(色谱级,默克两合股份有限公司),甲酸(分析级,国药集团化学试剂有限公司),磷酸(分析级,山东西亚化学工业有限公司),2, 4-二硝基苯肼盐酸盐(DNPH·HCl 分析级,东京化成工业株式会社),水为超纯水。
2 方法与结果
2.1 衍生条件
醛类常见的检测方法有气相色谱法、高效液相色谱法等[6]。由于硼替佐米对热不稳定,不宜采用气相色谱法检测。马庆国等[7]以 2, 4-二硝基苯肼衍生化试剂,采用高效液相色谱法检测甲醛,并探讨了衍生温度和缓冲液等对衍生化反应的影响;俞凌云等[8]以 2, 4-二硝基苯肼作为衍生化试剂,采用超高效液相色谱法检废水中乙二醛、甲醛等 7 种醛,具有良好回收率和灵敏度,同时专属性也比较好。本研究选择 2, 4-二硝基苯肼为衍生试剂,磷酸为酸催化剂,乙腈-水(20︰80,体积比)为溶剂,在室温条件下,衍生化 30 min 即得。(试验显示,衍生物峰面积 24 h 内无明显变化)。
2.2 色谱条件
色谱柱为 Waters symmetry C18(250 mm × 4.6 mm,5µm);以乙腈(体积分数0.1%甲酸)为流动相 A,以水(体积分数 0.1% 甲酸)为流动相 B,按表1进行线性梯度洗脱;流速为 1.0 mL•min-1;柱温为 30 ℃;检测波长为 364 nm;进样量为 5µL[9-10]。

Table 1 Linear gradient elution table表 1 线性梯度洗脱表
2.3 溶液的制备
2.3.1 2, 4-二硝基苯肼衍生化试剂溶液的制备
称取 2, 4-二硝基苯肼盐酸盐约 40 mg,置于 50 mL 量瓶中,加乙腈约 30 mL,再加磷酸 1 mL,超声使之溶解,用乙腈稀释至刻度,摇匀,即得。
2.3.2 异戊醛标准品溶液的制备
取异戊醛标准品约 30 mg,精密称定,置于 20 mL 量瓶中,加乙腈溶解并稀释至刻度,摇匀。精密量取 1 mL,置 50 mL 量瓶中,用乙腈稀释至刻度,摇匀,即得。
2.3.3 异戊醛-DNPH 对照品溶液的制备
精密量取异戊醛标准品溶液 1 mL,置于 20 mL 量瓶中,加乙腈 3 mL,再精密加入 2, 4-二硝基苯肼衍生化试剂溶液 1 mL,用水稀释至刻度,摇匀。室温放置 30 min,作为异戊醛-DNPH对照品溶液。
2.3.4 供试品溶液制备
取硼替佐米约 100 mg,精密称定,置于 20 mL 量瓶中,加乙腈 4 mL,水 1 mL,超声使溶解,精密加入 2, 4-二硝基苯肼衍生化试剂溶液 1 mL,用水稀释至刻度,摇匀。室温放置 30 min,作为供试品溶液。
2.3.5 空白溶液的制备
精密量取 2, 4-二硝基苯肼衍生化试剂溶液 1 mL,置于 20 mL 量瓶中,加乙腈 4 mL,用水稀释至刻度,摇匀,即得。
2.4 方法学考察
2.4.1 专属性试验
取本品约 100 mg,精密称定,置于 20 mL 量瓶中,精密加乙腈 3 mL,异戊醛标准品溶液 1 mL,2, 4-二硝基苯肼衍生化试剂溶液 1 mL 和水 1 mL,超声使溶解,用水稀释至刻度,摇匀,作为分离度溶液。取空白溶液,异戊醛-2, 4-二硝基苯肼对照品溶液,供试品溶液和分离度溶液,按“2.2”项方法检测,记录色谱图,见图 2。结果显示:空白溶液色谱图中在异戊醛-2, 4-二硝基苯肼峰位置无明显干扰峰,供试品溶液和分离度溶液色谱图中,异戊醛-2, 4-二硝基苯肼峰与相邻峰之间的分离度均大于 1.5,满足分离度要求,方法的专属性良好。
2.4.2 检测限和定量限考察
精密量取异戊醛-2, 4-二硝基苯肼对照品溶液 1 mL,置于 100 mL 量瓶中,加乙腈 25 mL,用水稀释至刻度,摇匀,作为定量限溶液。精密量取定量限溶液 2 mL,置 10 mL 量瓶中,加乙腈3 mL,用水稀释至刻度,摇匀,作为检测限溶液。分别量取定量限溶液和检测限溶液各 5µL 进样,记录色谱图。结果表明:异戊醛检测限为 0.016 ng,定量限为 0.080 ng,说明方法的灵敏度良好。
2.4.3 线性关系考察
按表2量取规定体积的异戊醛标准品溶液和 2, 4-二硝基苯肼衍生化试剂溶液,置于 20 mL量瓶中,用稀释剂稀释至刻度,摇匀。分别进样 5µL 测定,记录峰面积,以质量浓度ρ(mg•L-1)为横坐标,以峰面积(A)为纵坐标绘制标准曲线,得线性回归方程为A= 1.397 × 105ρ– 1.203 × 103,r=1.000 0。研究结果表明,异戊醛在质量浓度 0.015~2.998 mg•L-1范围内,线性关系良好。
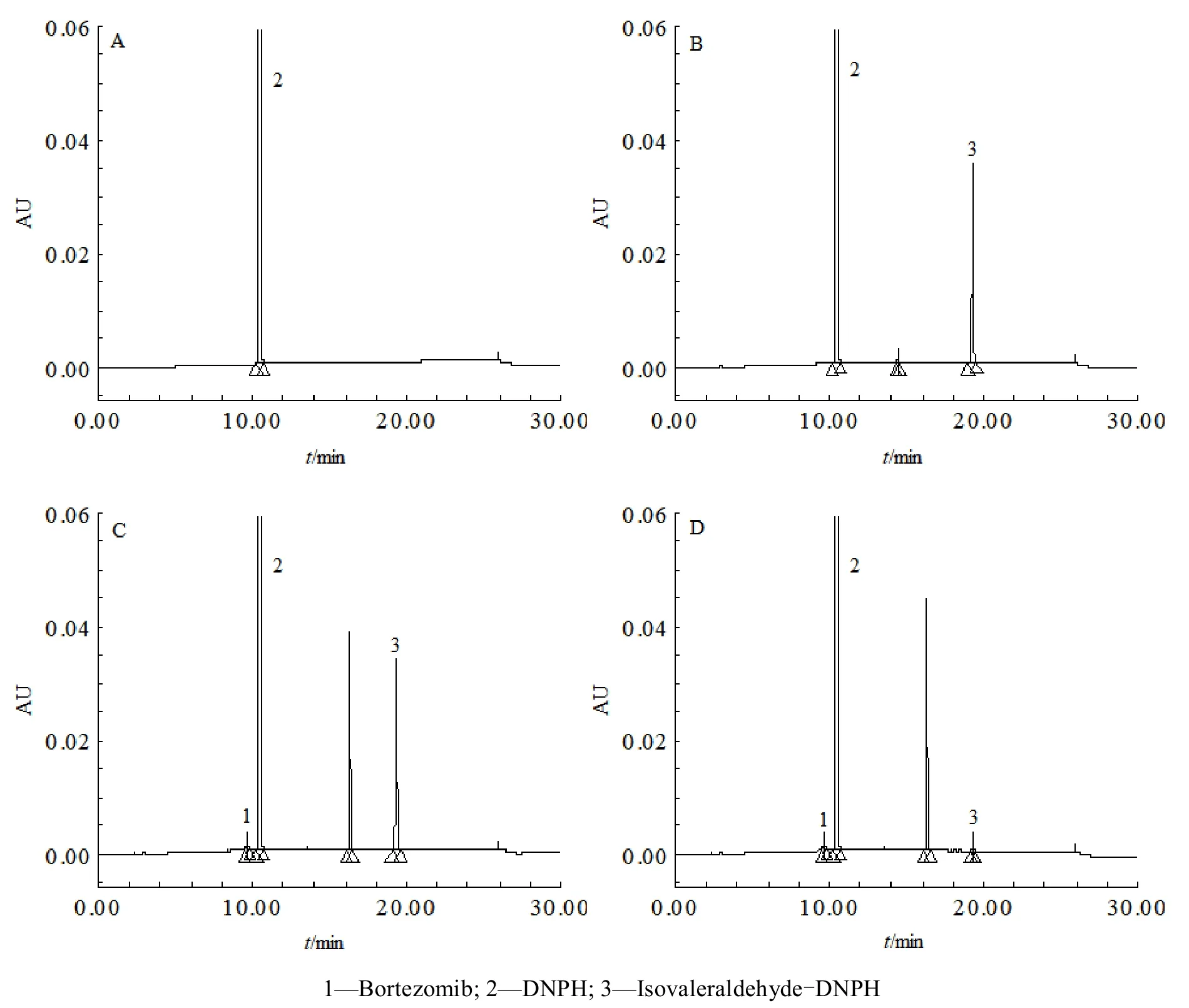
Fig. 2 HPLC chromatograms of blank (A), reference substances (B), resolution solutions (C) and samples (D)图 2 空白溶液(A),对照品溶液(B),分离度溶液(C)和供试品溶液(D)的HPLC色谱图
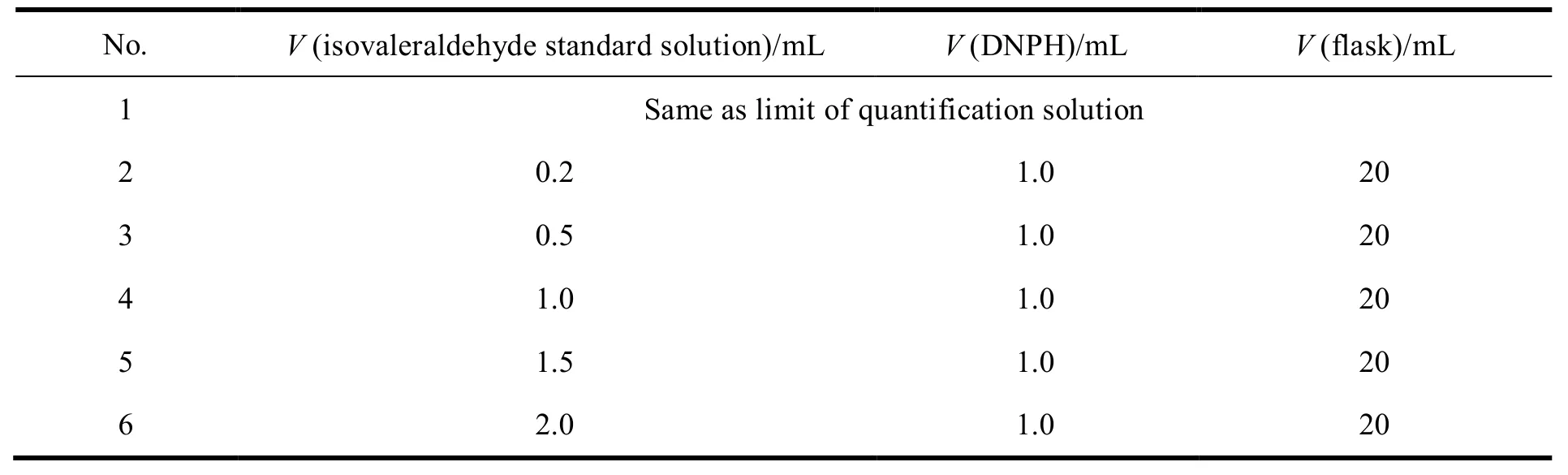
Table 2 Preparation of linear solutions表 2 线性溶液配制
2.4.4 精密度试验
取同一批次的硼替佐米,按“2.3.4”项下方法平行配制 6 份供试品溶液,按“2.1”项下色谱条件进样 5µL,记录供试品溶液中异戊醛-2, 4-二硝基苯肼的峰面积,按外标法以峰面积计算6 份供试品溶液中的异戊醛的含量。结果表明:6 份平行配制的供试品中异戊醛的含量的最大值为质量分数 0.000 7%,最小值为质量分数 0.000 6%,最大值与最小值之间的差值为 0.000 1%,表明方法重复性良好。
2.4.5 加样回收率试验
取同一批次的硼替佐米,精密称定 9 份,分成 3 组,每组 3 份,各组分别加入异戊醛标准品溶液适量(相当于限度浓度 0.03% 的 50%、100%、150%)和 2, 4-二硝基苯肼衍生化试剂溶液1 mL,加乙腈 3 mL,用水稀释至刻度。按“2.1”项下色谱条件进样,记录色谱图,计算异戊醛的回收率和 RSD,结果见表3,表明方法的回收率良好。
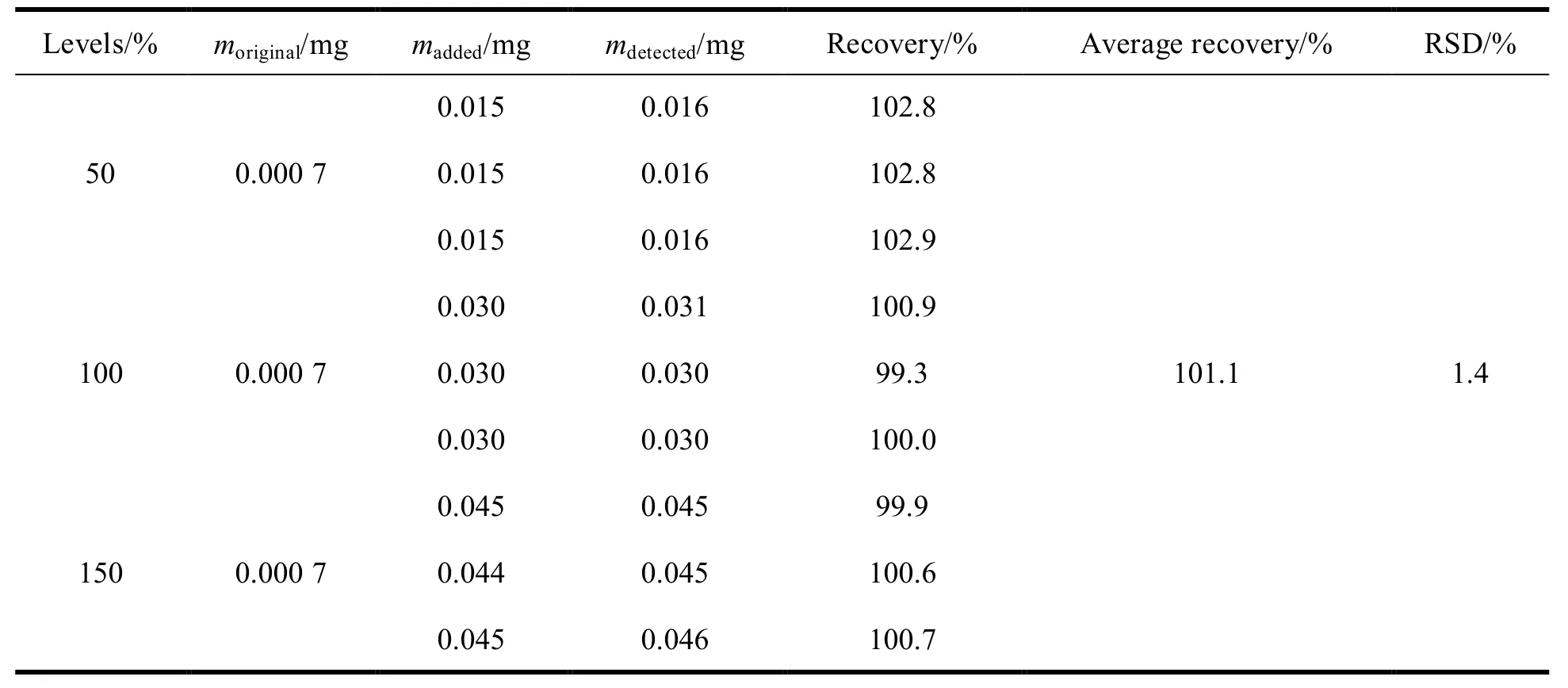
Table 3 Results of recovery test (n = 9)表 3 加标回收率结果(n = 9)
2.4.6 耐用性试验
精密量取对照品溶液和供试品溶液各 5µL,注入液相色谱仪,调节流动相流速(0.8 mL•min-1和1.2 mL•min-1)、柱温(25 ℃和35 ℃)、甲酸体积分数(0.09%和0.11%)和更换不同批号的色谱柱,记录色谱图。结果显示,各变化条件下系统适用性符合要求,与正常条件相比,供试品的异戊醛含量无明显差异,本方法耐用性良好。
3 结论与讨论
a. 由于异戊醛无明显紫外吸收,无法用液相直接测定,同时硼替佐米受高温不稳定,易降解产生异戊醛,因此亦不能采用气相色谱测定。本方法利用异戊醛与 2, 4-二硝基苯肼反应,生成稳定的腙类衍生物,建立了柱前衍生、高效液相色谱法测定硼替佐米中异戊醛含量的方法。
b. 该方法的灵敏度高,检测限可达到 0.016 ng,线性范围较宽,重复性好,异戊醛的回收率为 101.1%,耐用性好,可用于硼替佐米中异戊醛含量的测定。
