格列吡嗪控释片在健康人体的生物等效性预研究
2020-09-24李轶群王东凯
陈 清 ,李轶群,王东凯
(1. 沈阳药科大学 药学院,辽宁 沈阳 110016;2. 北京红林制药有限公司,北京101407)
格列吡嗪是第二代磺脲类口服降血糖药物[1],其作用机制是对胰岛β细胞的胰岛素分泌进行刺激,从而促进胰岛素的分泌。同时还可以增加脂肪细胞与胰岛素的结合,放大胰岛素的外周作用,减少肝糖的输出,目前临床广泛应用于 2 型糖尿病。本研究按照我国仿制药质量和疗效一致性评价的相关政策和要求,开展格列吡嗪控释片受试制剂与参比制剂的生物等效性预试验,由于北京红林制药有限公司首次注册时所采取的生物等效性试验为空腹给药,故本次预试验采取餐后给药的形式,为生物等效性正式试验提供数据支持和参考依据。同时,观察受试制剂与参比制剂在健康受试者中给药后的安全性。早期用高效液相色谱法测定血浆中格列吡嗪的浓度内源性干扰较多、灵敏度较低[2],尹然等建立的血浆中格列吡嗪的液相色谱-质谱联用法(high performance liquid chromatography-mass spectrometry,LC-MS)[3]采用大气辅助化学电离源(atmosphericpressure chemical ionization,APCI),选择性检测阳离子,具有较高的灵敏度和专属性。孙雪等建立的液相色谱串联质谱法(high performance liquid chromatography-tandem mass spectrometry,LC-MS/MS)采用电喷雾离子化源(electrospray ionization,ESI)电离方式[4],具有灵敏度高、精密度好、专属性强、快速简便等优点,适用于格列吡嗪的药代动力学及生物等效性的研究。故本文采用 LC-MS/MS 法测定血浆中格列吡嗪的血药浓度。
1 仪器与试药
1.1 仪器
QTRAP 5500 液质联用仪(美国ABSCIEX公司),1260 Infinity 高效液相色谱仪(美国 Agilent公司)。
1.2 药品与试剂
受试制剂(T):格列吡嗪控释片(北京红林制药有限公司,国药准字 H20084634,规格为5 mg/片,批号 21701020),参比制剂(R):格列吡嗪控释片(Pfizer Pharmaceuticals LLC,辉瑞制药有限公司进口分装,进口药品注册证号 H20100239、H20100240,国药准字 J20100129,规格为5 mg/片,批号 S24038),格列吡嗪对照品:TLC Pharmaceutical Standards(质量分数为99.1%,批号 1169-074A1),内标 Glipizide-d11,Toronto Research Chemicals INC(质量分数为98.0%,同位素质量分数为 97.6%,批号 7-SDJ-115-1)。
2 方法
2.1 受试者筛选
在第 1 次给药 -14~-1 天对中国健康志愿者进行筛选,本研究经伦理委员会及中国人类遗传资源管理办公室批准,健康志愿者对本研究知情,并签署知情同意书。经筛选,共 12 名健康受试者正式入组(男女随机,其中男性 7 例,女性 5 例)。所有受试者于试验前接受全面体检,结果正常。受试者均无既往病史和药物过敏史,精神状态良好,无吸烟、酗酒嗜好,试验前两周未服用任何药物。
2.2 分组及给药
采用开放、随机、两周期交叉试验设计,将 12 例中国健康受试者随机分为 2 组,每组 6 例。受试者于给药前一天禁食过夜 10 h 以上后,于第 1 天餐后口服受试制剂或参比制剂 1 片,用240 mL 质量分数为 20% 葡萄糖水送服。于给药前 60 min 内(0 h)和给药后 1、2、3、4、6、8、9、10、12、16、24、36、48、72 h 由静脉取血 4 mL,血样置含乙二胺四乙酸二钾抗凝剂离心试管中,轻柔颠倒混匀,离心((4 ± 2)℃,4 000 rpm,10 min),分离后的血浆于(–80 ± 10)℃冷冻保存,待测。清洗期 7 天后交叉给药。
2.3 血浆样品分析方法的建立
2.3.1 血浆样品处理方法
精密量取 100µL 血浆样品至 2 mL 离心管中,加入 400µL 乙腈,涡旋混合 5 min,于 4 700 rpm、4 ℃ 离心 10 min,取上清液 100µL,向其中加入 100µL 体积分数为 10% 的乙腈稀释,涡旋混合 5 min,准备进样分析。
2.3.2 色谱条件与质谱条件
色谱条件:色谱柱为 Kinetex C18(50 mm ×2.1 mm,2.6µm);流动相 A:体积分数为 0.1%甲酸,流动相 B:乙腈;洗脱方式:梯度洗脱(0.6 min、体积分数为65%的B;1.2 min、体积分数为 95% 的 B;2.0 min、体积分数为 50% 的 B);流速:0.6 mL•min-1。
质谱条件:电喷雾离子化源(electrospray ionization,ESI),正离子模式,多反应监测(multiple reaction monitoring,MRM),离子化电压:5 500 V,离子源温度:550 ℃,碰撞能量(collision energy,CE)19 V。
2.4 分析方法的确证
2.4.1 专属性
在 2.3.2 条件下格列吡嗪和内标(internal standard,IS)的出峰时间分别在 0.16 min 和 0.37 min左右,空白血浆在两者的峰位处均无杂质干扰,专属性良好(见图1)。
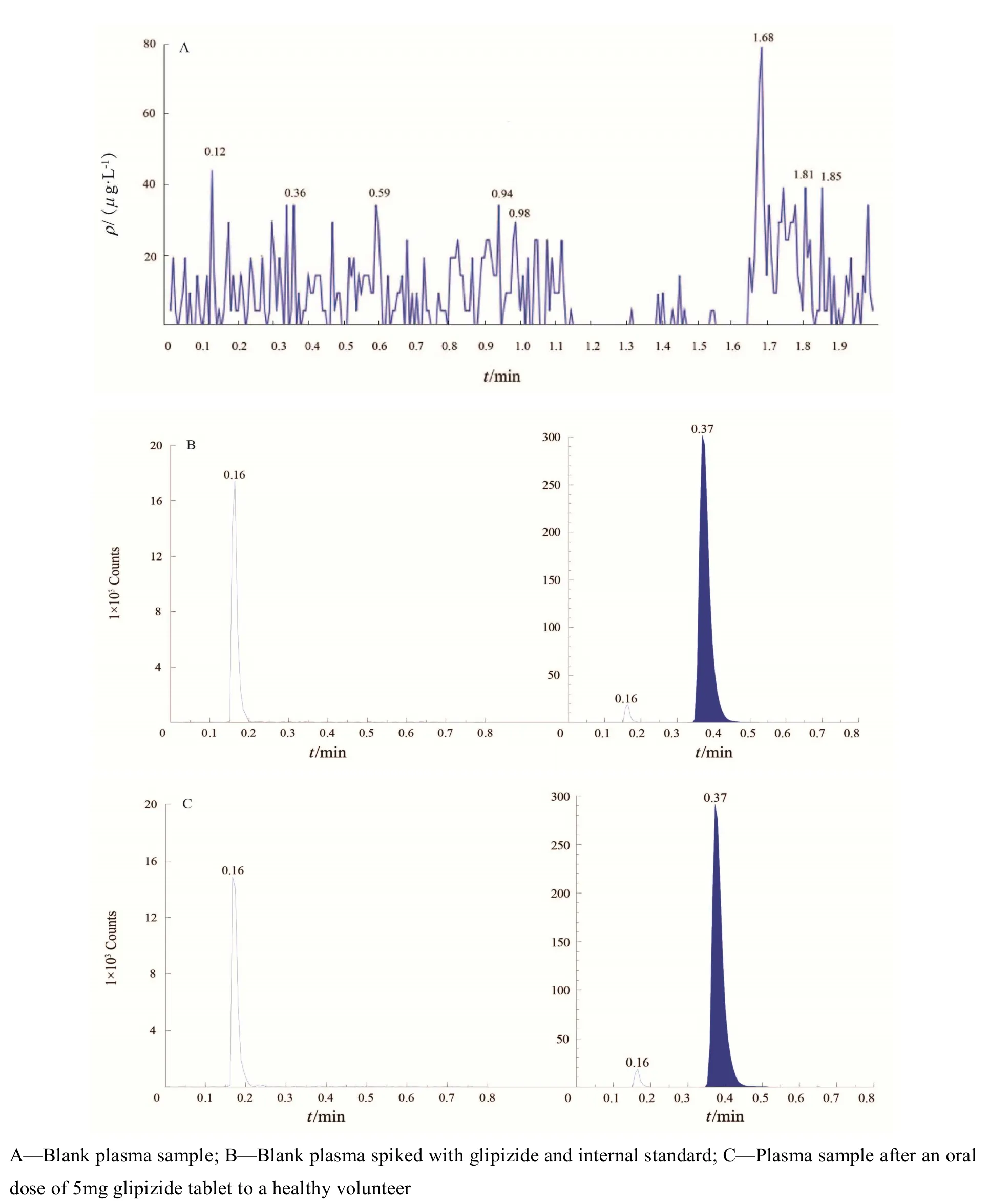
Fig. 1 Typical chromatograms of glipizide and internal standard图 1 血浆中格列吡嗪色谱图
2.4.2 标准曲线及定量下限
配制血药质量浓度为 2.00、4.00、20.0、50.0、100、150、250、300µg•L-1的标准曲线样品,进样检测,标准曲线回归方程为Y= 1.046×10-2X- 4.000×10-5(r= 0.999 6)。结果表明,格列吡嗪血药浓度在 2.00~300µg•L-1范围内线性关系良好,定量下限为 2.00µg•L-1。
2.4.3 精密度和准确度
处理定量下限、低、中、高 4 个质量浓度(2.00、6.00、40.0、200µg•L-1)的血浆样品各 6 份,进样 3 天,批内精密度 RSD 为 0.7%~4.8%,批内准确度均值(与标示浓度的偏差)范围-5.0%~4.7%;批间精密度 RSD 为 2.0%~4.9%,批间准确度均值(与标示浓度的偏差)范围-1.3%~1.5%。符合生物样本分析检测要求。
2.4.4 提取回收率
配制质量浓度分别为 6.00、40.0、200µg•L-1的血浆样品各 6 份,三个质量浓度的格列吡嗪提取回收率分别为(98.8 ± 3.4)%、(111.7 ± 8.2)%和(111.7 ± 3.7)%,平均提取回收率为(107.4± 5.1)%。符合生物样本分析检测要求。
2.5 统计学方法
采用 WinNonlin 7.0 软件用非房室数学模型分析方法,进行药动学参数的估算和分析。对AUC 和ρmax作对数转换后进行方差分析,用双单侧t检验和计算 90% 置信区间的统计分析方法进行等效性检验。若ρmax、AUC0-t和 AUC0-∞的几何均值比的 90% 置信区间在 80.00%~125.00% 等效区间内,即可判定两制剂人体生物等效。
3 结果
3.1 受试者入组及完成情况
实际入组 12 例受试者,均完成试验。纳入药动学评价 12 例。
3.2 药-时曲线
受试者餐后服用格列吡嗪受试制剂和参比制剂平均血药浓度-时间曲线,见图2。
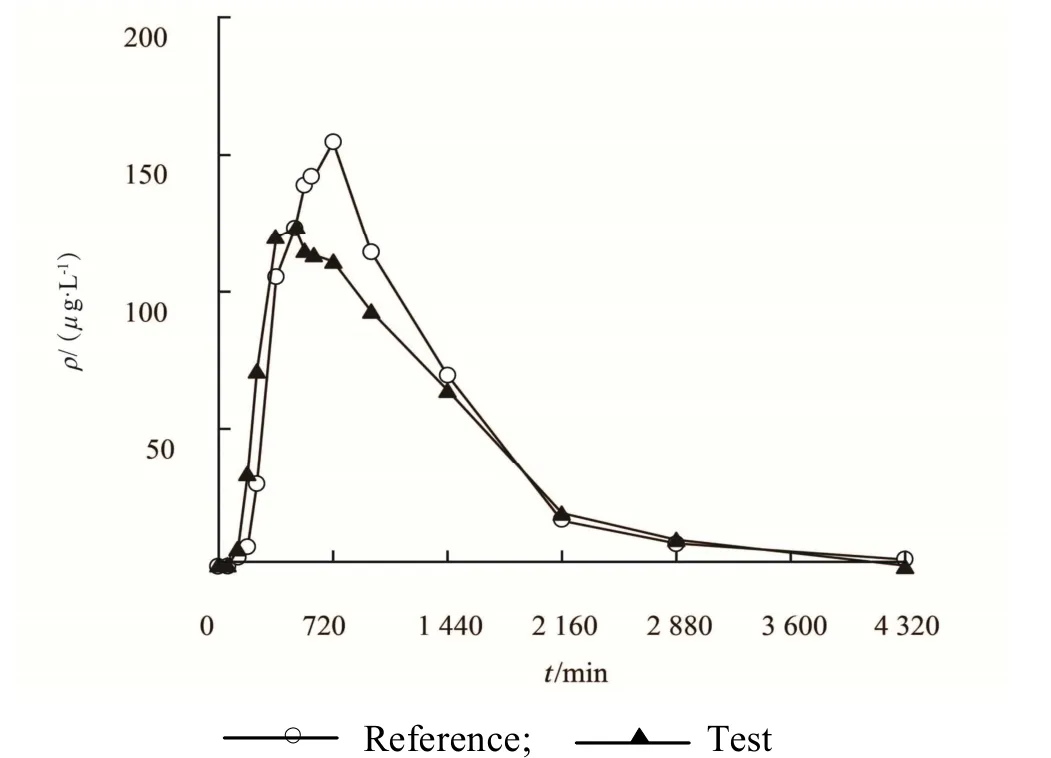
Fig. 2 Mean plasma concentration-time curves of glipizide after taking the test and reference drugs in fed state图 2 受试者餐后口服格列吡嗪受试制剂和参比制剂后的平均血药浓度-时间曲线
3.3 药代动力学参数
12名受试者分别单次口服参比制剂与受试制剂后的药动学参数见表1、表2。
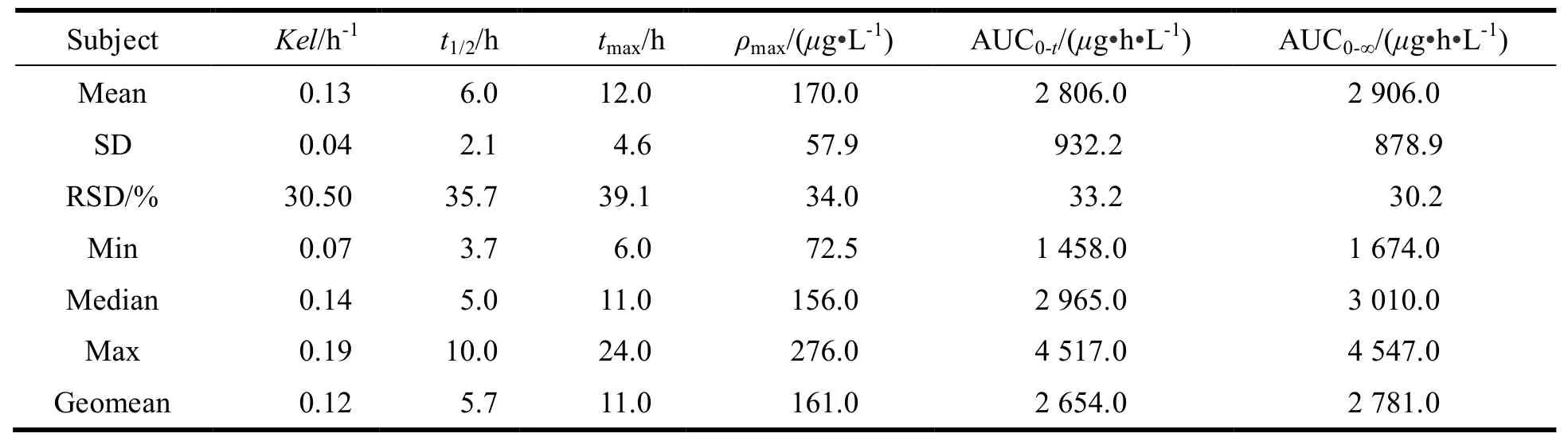
Table 1 The main pharmacokmetic parameters of glipizide after taking the reference drugs in 12 healthy volunteers表 1 12名受试者口服参比制剂的药动学参数
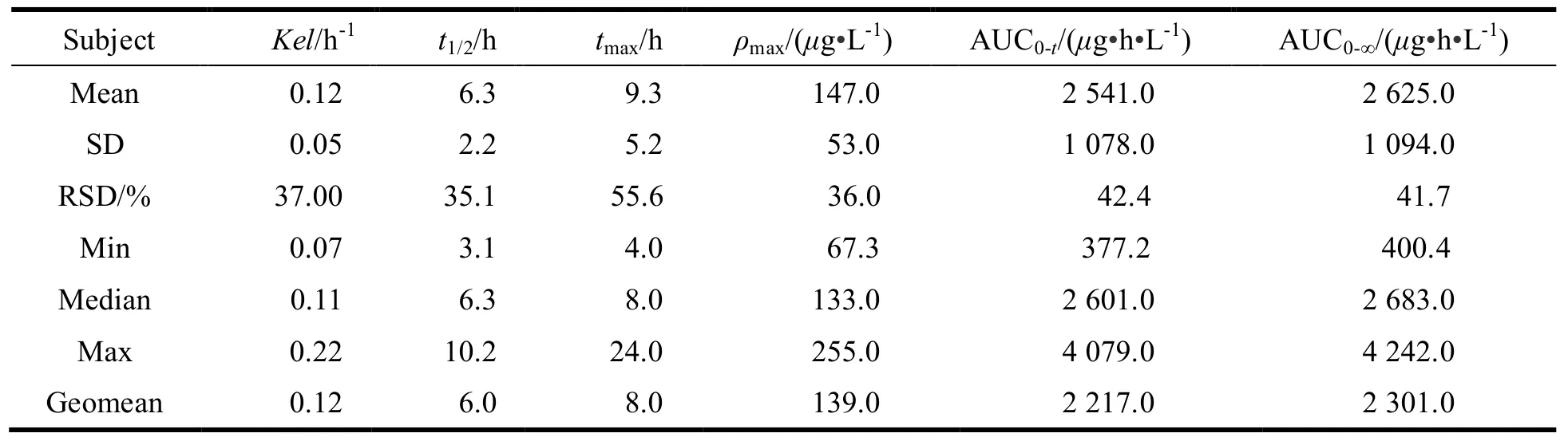
Table 2 The main pharmacokmetic parameters of glipizide after taking the test drugs in 12 healthy volunteers表 2 12名受试者口服受试制剂的药动学参数
3.4 生物等效性检验
ρmax、AUC0-t及 AUC0-∞在进行对数转换后进行双向单侧t检验。计算受试制剂与参比制剂ρmax、AUC0-t和 AUC0-∞的几何均值比的 90% 置信区间。表3为生物等效性评价的统计结果。

Table 3 The evaluation of bioequivalence in 12 healthy volunteers表 3 12名受试者生物等效性评价的结果
受试制剂与参比制剂的ρmax的几何均值比的 90% 置信区间为(71.76%~103.73%),AUC0-t的几何均值比的 90% 置信区间为(62.07%~112.37%),AUC0-∞的几何均值比的 90% 置信区间为(61.91%~110.57%),均超出 80.00%~125.00%,格列吡嗪控释片的受试制剂和参比制剂不符合生物等效性标准。
3.5 方差分析
采用 WinNonlin 7.0 软件进行统计分析,格列吡嗪的ρmax、AUC0-t及 AUC0-∞再进行对数转换后,采用交叉试验设计的方差分析,分析给药序列间(sequence)、制剂间(formulation)、个体间(sequence × subject)和周期间(period)的差异有无统计学意义(P> 0.05,表示差异无统计学意义)。方差分析结果见表4~表6。结果表明:对于ρmax、AUC0-t、AUC0-∞,给药序列间、个体间、制剂间和周期间的差异均无统计学意义(P> 0.05)。
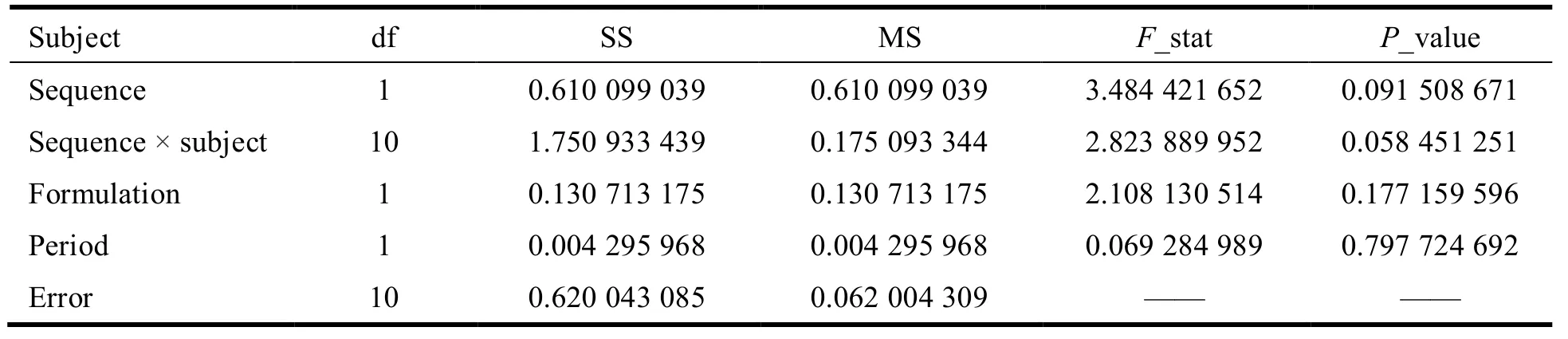
Table 4 Variance analysis of ln ρmax表 4 ln ρmax的方差分析结果
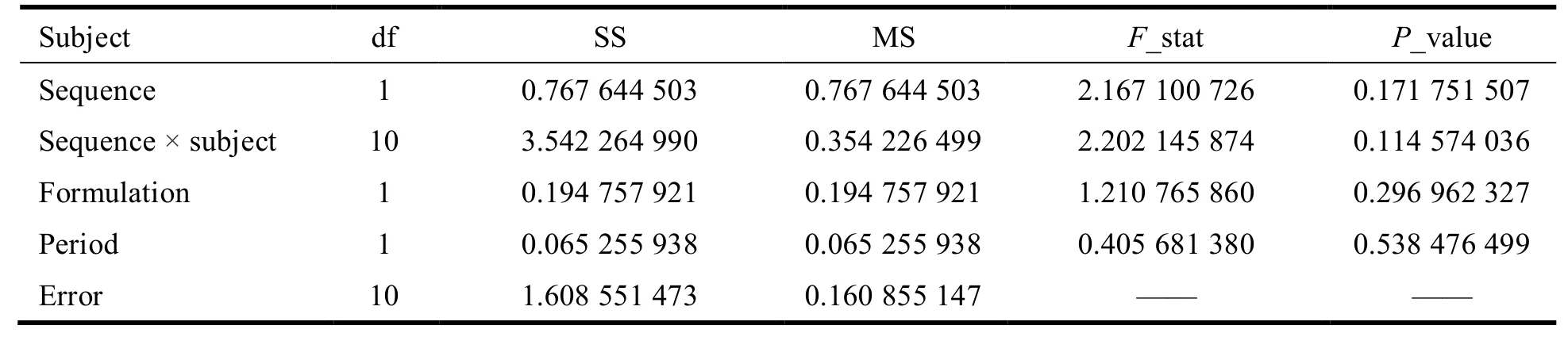
Table 5 Variance analysis of ln AUC0-t表5 ln AUC0-t的方差分析结果
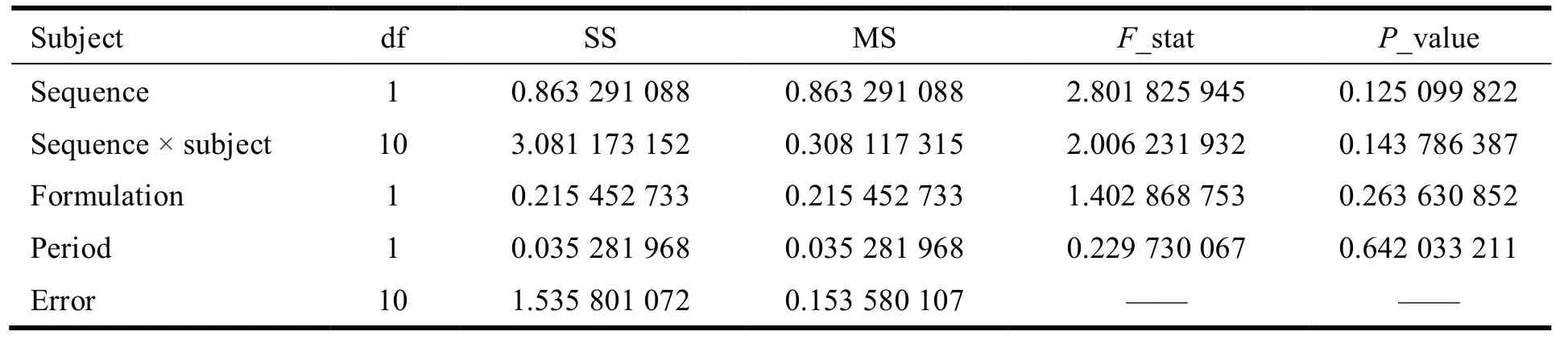
Table 6 Variance analysis of ln AUC0-∞表 6 ln AUC0-∞的方差分析结果
3.6 安全性评价
试验中,12 例健康受试者全部完成试验,6 例受试者共发生 7 例次不良反应。其中受试制剂有 4 例(33%)受试者 4 例次,参比制剂有 3 例(25%)受试者 3 例次。停药后或对症治疗后,均已随访至恢复,整个试验过程中无严重不良事件发生。
4 讨论
从受试者餐后服用格列吡嗪受试制剂和参比制剂的药-时曲线来看,参比制剂血药浓度较受试制剂高,可能是由于参比制剂所用的聚氧乙烯与受试制剂所用的聚维酮在调节药物释放速率上存在一定差异[5]。聚氧乙烯是一种结晶性、热塑性的水溶性聚合物,由于其存在 C-O-C 键,通常具有柔顺性,其吸水速度和水合速度均较慢,因此药物释放的时滞较长。聚维酮是非离子型水溶性高分子化合物,极易溶于水,以聚维酮为载体的控释片通常具有较短的时滞,因此受试制剂的tmax比参比制剂快了约 3 个小时。但聚维酮吸水膨胀后可能形成比较致密的凝胶层,又进一步阻滞了药物的溶出。提示下一步一致性评价研究,应从调节药物释放的辅料特性着手,优化其处方,适当调整处方比例,并充分考察各因素影响。另本次试验为预试验,由于预试验样本量较小,个体内变异系数较高,个体的数据对最终结果影响较大。提示在正式生物等效性试验时,可以根据本次预试验ρmax的个体内变异系数估算扩大样本量,以期达到和原研药生物等效的目的。
