毛细管电泳用于药物分析的研究进展
2020-09-18许旭,陈钢,刘浩
许 旭, 陈 钢, 刘 浩
(1. 上海应用技术大学, 上海 201418; 2. 上海市食品药品检验所, 上海 201203; 3. 国家药监局治疗类单抗质量控制重点实验室, 上海 201203)
药物分析是毛细管电泳(CE)的重要应用领域,持续提供着许多不同类型和有实际研究价值的研究需求,也不断将CE的研究成果应用到药物研发、生产以及临床应用与评价等各环节。CE用于药物分析的研究文献较多,本文从药品分析领域中的化学药物、中药、生物制品,以及体内药物分析几个方面综述了近几年毛细管电泳在这些传统药物分析中应用的研究进展。对于现代药物分析研究比较活跃的理化常数测定、亲和毛细管电泳与结合常数研究(包括药物与受体间的相互作用等)、临床分析生物标志物、代谢组学,以及微流控芯片CE分析等方面的研究,文献内容较多,本文限于篇幅未做讨论。本文主要涉及2017年1月至2020年2月近3年的文献,也包括一些2016年的文献和之前的少量相关文献。在概述这段时期CE在传统药物分析领域新进展的同时,本文讨论了目前这些传统药物分析领域的需求,以及CE在其中的地位、挑战和机遇。
Deeb等[1]在对2013~2015年CE在药物分析中的应用研究评述中,提到不少传统药物分析领域的技术需求,包括人用药品注册技术要求国际协调会(ICH)对药物分析方法的要求,以及进样方法和检测灵敏度等常见CE方法问题。屈锋实验室[2-5]近3年以及之前的毛细管电泳年度综述均包括药物分析的内容。Kristoff等[6]总结了CE在蛋白质、药物、糖基化分析,以及外泌体与病毒分析等生物分析方面的重要进展。Voeten等[7]发表在AnalyticalChemistry的年度综述总结了CE在2015年9月至2017年9月的研究进展。近期有关CE-MS的研究引起了人们的关注,Stolz等[8]综述了CE-MS的研究进展,包括在单克隆抗体与药物的应用。周韦等[9]综述了CE-MS在药物和生物制品分析中的应用。本文从小分子药物及其有关物质、手性分离、中药与天然产物、体内药物分析、大分子与生物制品分析几个方面具体讨论近几年CE在这些传统药物分析中应用的研究进展。
1 小分子药物及其有关物质
1.1 法规需求
小分子药物的分析越来越关注到ICH的方法要求。Deeb等[1]在评述时关注到ICH Q8指南中“质量源于设计”(quality by design, QbD)的策略对药物分析方法建立的要求。戴胜云等[10]综述了QbD在药物分析方法开发中的应用研究进展。QbD要面向需求,通过系统化、结构化、层层递进的方式建立分析方法。Pasquini等[11]基于QbD原理,使用γ-环糊精-正丁醇-胆酸钠-硼酸盐分离缓冲液,建立了毛细管胶束电动色谱(MEKC)快速测定卡托普利、氢氯噻嗪及其有关物质的方法。
中国药典四部(2015版)在通则9101“药品质量标准分析方法验证指导原则”中规定[12]:精密度可接受范围与样品中待测定成分含量有关,待测定成分含量在1%以上时重复性(RSD)要求低于2%,而CE直接定量的重复性(RSD)多在3%左右。虽然不少文献中日内RSD小于2%,但推广应用时常常达不到要求。因此,对多数含量高于1%的药品主成分,CE方法需要改进重复性问题。郭怀忠等[13]曾经在2005年系统讨论过影响毛细管电泳分析结果重现性的因素及其控制途径,至今仍然有很好的参考价值。改进精密度的常见方法是内标法。Lago等[14]以呋喃苯胺酸为内标,建立了测定替帕那韦(tipranavir)胶囊的毛细管区带电泳法(CZE),方法精密度<2%,将该法用于药品在酸性、碱性、热性、光解性和氧化性条件下的稳定性评价,显示氧化为主要降解途径。Guichard等[15]建立了两种CE方法用于内标法定量分析注射剂中的16种抗肿瘤药物。一种是使用含50%乙腈的分离缓冲液建立的CZE方法,用于分析阿霉素、表阿霉素、伊达比星、道诺霉素、伊立替康、拓扑替康、长春新碱、长春地辛、长春碱和长春瑞滨。另一种是MEKC使用含20%乙腈的硼酸盐-十二烷基硫酸钠(SDS)分离缓冲液测定甲氨蝶呤、培美曲塞、依托泊苷、磷酸依托泊苷、磷酸氟达拉滨和5-氟尿嘧啶。胡雯雯等[16]使用阎超研究团队[17]此前开发的基于定量阀进样的商品化仪器(原理图见图1),同时测定复合维生素B片中5种维生素,直接测定峰面积的日内RSD在1.9%以内,显著优于普通CE仪器。近期另一台值得关注的仪器是浙江大学方群教授实验室[18]开发的手持式CE仪器,使用激光诱导荧光检测、短柱和缺口管进样技术,具有体积小、成本低的特点,已用于氨基酸、蛋白质和DNA的分析。
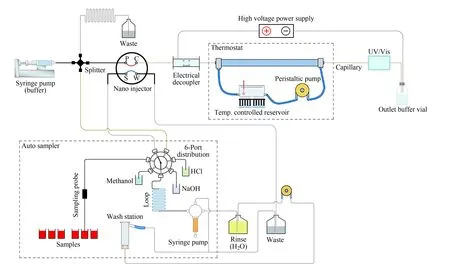
图 1 基于定量阀进样的高精度毛细管电泳仪器原理图[17]Fig. 1 Schematic overview of automated quantitative capillary electrophoresis system[17]
1.2 电容耦合非接触电导检测法(C4D)
C4D适用于缺乏生色团成分的检测,近来得到广泛应用。Elbashir等[19]综述了2014年2月到2016年10月期间CE-C4D在药物、生物医学和食品分析领域的应用文献,其中以药物分析的应用居多。Cunha等[20]建立了在2 min内快速测定不同制剂中可待因、邻甲苯海明、异丙嗪、东莨菪碱、曲马多和扑热息痛含量的CE-C4D方法,检出限为0.62~15 μmol/L。de Castro Costa等[21]采用CE-C4D在1 min内超快速同时测定了药品中铵和苯海拉明的含量。他们使用2-(N-吗啉诺)乙醇磺酸-氢氧化锂(pH 6.0)作为背景缓冲液。氨和苯海拉明的检出限分别为0.04 mmol/L和0.02 mmol/L。他们[22]还提出CE-UV和CE-C4D快速测定精氨酸、抗坏血酸和天冬氨酸的方法,每小时分别可以测定23和78次,LOD均为0.01~0.04 mmol/L。Nguyen等[23]设计了一种CE-C4D系统,可用于β-内酰胺抗生素的质量控制与掺伪识别。Evans等[24]用便携式CE-双C4D分别测定了3种含伪麻黄碱的市售片剂(见图2)和两种违禁药物(甲基酮和对甲氧基甲基苯丙胺)的无机阴离子和无机阳离子谱。10个阳离子的检出限为0.10~1.25 μmol/L, 8个阴离子检出限为0.13~1.03 μmol/L。两类离子的CE分离分别都在6 min内完成,测得的离子指纹数据可用于对未知样品的分类。
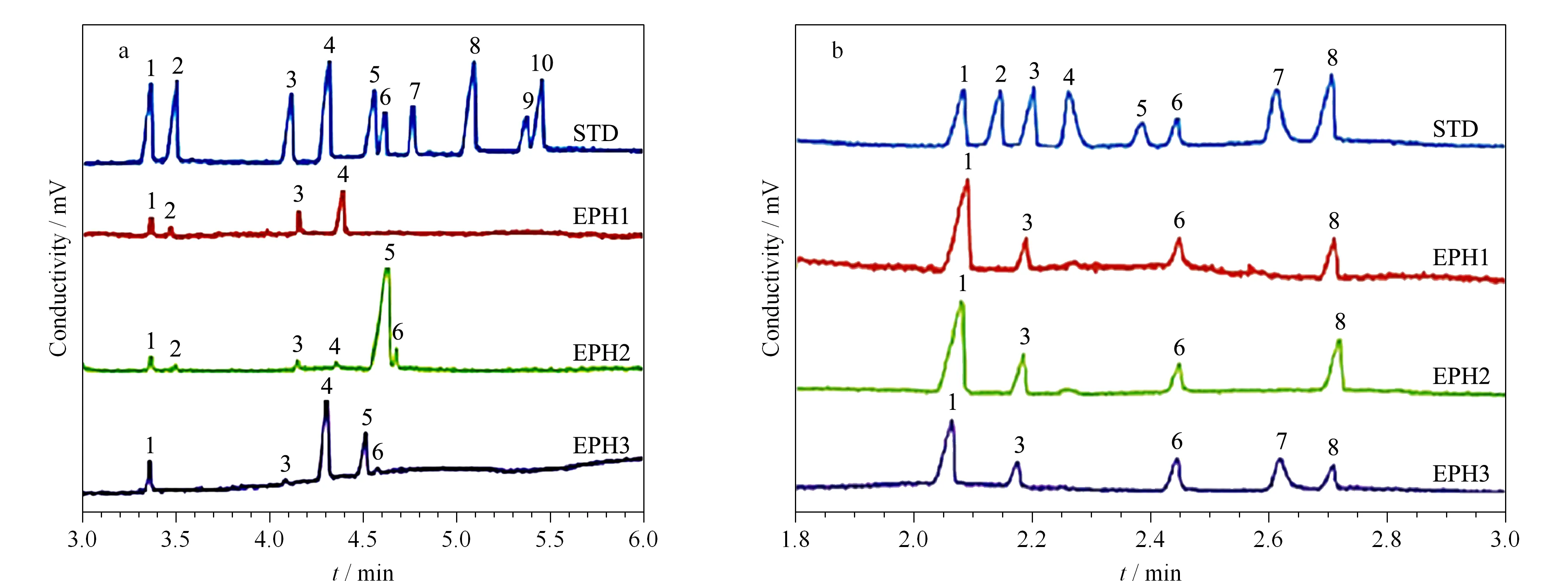
图 2 3种市售含伪麻黄碱药片的(a)无机阳离子和(b)阴离子CE-C4D谱图[24]Fig. 2 CE-C4D chromatograms of (a) cations and (b) anions in the three kinds of commercially available tablets containing pseudoephedrine[24] C4D: capacitively coupled contactless conductivity detection; STD: standard samples; EPH: pharmaceutical tablet containing pseudoephedrine. Peaks in Fig. 1a: 1. Cs+ (IS), 2. 3. K+, 4. Ca2+, 5. Na+, 6. Mg2+, 7. Mn2+, 8. Sr2+, 9. Ba2+, 10. Li+; peaks in Fig. 1b: 1. Cl-, 2. 6. F-(IS), 7.
1.3 间接检测方法
1.4 CE-MS分析
dos Santos等[28]用CE-MS/MS测定天然减肥药和膳食补充剂中安非他明及其衍生物芬特明(PTM)、甲基苯丙胺(MAM)、甲二氧基安非他明(MDA)、甲二氧基甲基安非他明(MDMA)和甲二氧基乙基安非他明(MDEA)。样品经改良的QuEChERS方法处理,检出限为0.02~0.06 μg/L。Ouyang等[29]采用电渗流泵鞘流纳喷接口,实现了负离子模式下的毛细管等电聚焦(cIEF)-MS联用,并用于按照电荷差异精细分离识别硫酸软骨素和硫酸肝素中不同结构的酸性低聚糖,鞘流液是含乙酸铵的甲醇-水溶液。此前该团队的Sun等[30]采用相同的电渗流泵鞘流接口,用CZE-MS在正离子模式下分析了肝素低聚糖和低分子量肝素,使用的分离缓冲液是挥发性碳酸氢铵,鞘流液是含甲酸的甲醇-水溶液。Ansar等[31]先用含10%蔗糖的磷酸缓冲液(20 mmol/L, pH 6.5),在对脂质体最小影响的条件下,用CE直接测定脂质体阿霉素制剂中未包封阿霉素的含量。他们[32]还进一步建立了CE与电感耦合等离子体(ICP)-MS/MS联用方法,可在脂质体破坏最小的情况下直接定量测定阿霉素脂质体制剂中在脂质体内外不同状态的硫酸盐含量。
1.5 离子液体
李敏等[33]以30 mmol/L的1-丁基-3-甲基咪唑四氟硼酸盐离子液体水溶液为背景电解质,以头孢拉定为内标,CE同时测定头孢噻肟、头孢唑啉、头孢曲松和头孢米诺4种注射用头孢药物的含量。离子液体经二氯甲烷萃取回收可重复使用。离子液体在手性药物分析方面也有较多研究[34,35]。
1.6 药物有关物质分析
国家药品监督管理局药品审评中心公布的《化学药物杂质研究的技术指导原则》[36]指出,“由于各种分析方法均具有一定的局限性,因此在进行杂质分析时,应注意不同原理的分析方法间的相互补充与验证,如HPLC与TLC及HPLC与CE的互相补充……”。说明在药品杂质分析研究时,需要用CE作为“互补技术”去发现目前HPLC方法难以分离分析甚至未检出的杂质。这种“互补技术”的应用也是各国药品审评指导原则的要求。
药品中诸杂质的种类与含量被总称为杂质谱,Holm等[37]综述了药物杂质谱分析的研究进展,讨论了ICH在药物杂质风险控制方面的若干准则和策略。Dispas等[38]介绍了可应用于杂质检测方法的QbD原理和统计策略,并对QbD应用这类分析方法的文献进行了综述。Görög[39]则评述了包括CE在内的近十年来对映体杂质的药物杂质谱和降解谱方面的研究进展。毕萌萌等[40]建立了原料药中呋喃西林及其杂质5-硝基糠醛二乙酯的MEKC分析方法。杨直等[41]建立了CZE测定盐酸雷尼替丁注射液中雷尼替丁和3个有关物质含量的方法。Fayed等[42]在化学计量学优化的基础上建立了CZE同时测定佐芬普利钙、氢氯噻嗪,以及氢氯噻嗪的两种主要杂质氯噻嗪(chlorothiazide)和沙胺(salamide)。de Souza等[43]建立了CE分析呋喃苯胺酸(furosemide)及其降解样品的方法,并做了方法认证,该药在中性、酸性和碱性条件下的水解,以及氧化、热和光引起的降解产物不影响其定量。
1.7 手性药物的对映体杂质分析
中国药典四部(2015版)[12]在通则9102“药品杂质分析指导原则”指出,对于立体异构体杂质的检测广泛采用手性色谱法和高效毛细管电泳法。
Saz等[44]综述了环糊精用于CE手性药物分析的研究进展。Fanali等[45]在详细总结CE手性拆分各方面已有成果的基础上,讨论了近十年进展缓慢的原因和未来发展趋势。潘聪洁等[46]综述了2013~2015年CE用于手性分离的进展。刘明霞等[47]综述了2017~2019年CE在手性分离分析方面的进展。杜迎翔等[48]综述了近年来二元手性选择剂CE拆分体系的进展。吐尔逊·贾娜尔[34]综述了手性离子液体在毛细管电泳手性分离中的应用。Hancu等[49]综述了CE在抗抑郁药(氟西汀、西酞普兰、舍曲林、文拉法辛和度洛西汀)对映体分离中的应用。Ali等[50]综述了手性喹诺酮类药物对映体拆分的色谱和电泳方法,还提出一些选择手性分离条件的建议。
本文侧重讨论与传统药物分析应用领域密切相关的研究进展,不再特别讨论手性拆分剂与拆分方法研究方面的内容。
1.7.1检测灵敏度
对映体杂质分析通常需要在手性分离的基础上能够定量含量低至0.1%的对映体,《化学药物杂质研究的技术指导原则》[36]对不同剂量的原料药和制剂中杂质(包括对映体杂质)的报告限度一般为0.03%~0.1%,质控限度一般为0.05%~1.0%,这对CE对映体分离的检测灵敏度提出了一定要求。Sánchez-López等[51]综述了2013年6月到2015年5月有关手性CE中改进检测灵敏度的进展。esták等[52]以消旋美沙酮为样品,带负电荷的β-环糊精硫酸酯为手性选择剂,通过对低电导率样品溶液中的阳离子进行柱头场放大浓缩,改善CE手性分析的检测灵敏度。Casado等[53]比较不同环糊精的实验结果,结果硫酸化-γ-CD(sulfated-γ-CD)作为手性拆分剂分离伊伐布雷定(ivabradine)效果最好,而且加入氨基酸手性离子液体(单独使用没有得到手性分离)则可以增加分离度,建立的方法可以检测0.1%的对映体杂质。徐梓馥等[54]用羧甲基-β-CD和L-组氨酸-Cu2+双手性手性选择剂CE拆分氧氟沙星对映体,可以检测到0.1%的对映体杂质成分。
1.7.2QbD原理应用
基于QbD原理,Orlandini等[55]建立了环糊精修饰的胶束电动色谱法同时测定手性药物氨布里西坦(ambrisentan)的对映体杂质和有关物质。Pasquini等[56]使用羟丙基-γ-环糊精(HP-γ-CD)手性拆分,建立了CE同时测定手性药物西那卡塞(cinacalcet)对映体纯度和杂质含量的方法。基于QbD,以手性分离度和分析时间为指标,经过Box-Behnken设计、确定“可操作的方法设计区域(method operable design region, MODR)”、Plackett-Burman耐用性检验,以及基于ICH的方法验证,最后用于实际样品分析。Krait等[57]基于QbD,以硫酸化β-CD为拆分剂建立了CE测定右美托咪定手性纯度的方法,可测定0.1%的杂质含量。
1.7.3体液手性药物分析
生物液体中药物低浓度的手性分离分析是一项具有挑战性的任务。ebestová等[58]利用CE-ICP/MS以sulfated-γ-CD作为手性拆分剂测定了尿样中的抗肿瘤药奥沙利铂对映体。检出限为64 ng/mL。Wu等[59]用聚去甲肾上腺素包被磁性纳米颗粒和手性毛细管电泳手性分离分析了3种β-受体拮抗剂(卡特洛尔、美托洛尔和倍他索洛尔)。结合场增强进样,检出限为0.401~1.59 ng/mL,并用于人体尿液样品分析。该实验室Xiao等[60]还用聚多巴改性磁性纳米颗粒固相萃取结合手性CE方法,手性分离分析了尿样和牛血清中微量加标氧氟沙星外消旋体。结合压力辅助场增强进样,检出限低至0.29 ng/mL。杨四梅等[35]使用L-脯氨酸-Cu2+-氯化-1-丁基-3-甲基咪唑([BMIM]Cl)的Tris-磷酸缓冲液,以离子液体促进的手性配体交换CE分离分析去氧肾上腺素光学异构体,并用于加标血液和尿液样品中R和S型去氧肾上腺素的测定。
手性CE分析可以看成是特殊的难分离杂质分析,与化学药物杂质研究的需求相关。目前使用手性固定相的HPLC应用较多,也解决了大部分对映体杂质的分析检测问题。CE因其易于在缓冲液中使用不同的手性添加剂而独具特色,可在手性HPLC难分离对映体样品的分析方面发挥重要作用。而且CE手性拆分时,拆分剂与对映体药物处于均处于自由溶液中,为研究手性药物与受体的识别提供了良好条件。
1.8 制剂分析
Contin等[61]建立了MEKC分析儿科制剂中艾地苯醌(idebenone)的分析方法,并做了方法认证。结果表明,该成分存在两种不同氧化还原状态和生物活性。Kogawa等[62]提出一种定量分析片剂中利福昔明(rifaximin)的CE方法。王萍等[63]用CE测定消毒剂与药品中的利巴韦林时,利用十六烷基三甲基溴化铵反转电渗流缩短了分析时间。Junger等[64]建立了CE快速测定不同制剂中麻醉剂苯佐卡因和利多卡因的方法。Mufusama等[65]建立了CZE测定抗疟复方制剂中阿莫地喹及其中含量≤0.5%的3种合成杂质的方法,并根据ICH的质量指南Q2(R1)文件的规定做了方法验证。
1.9 其他
Kowalski等[66]利用SDS单体与大环抗生素的相互作用,结合场放大进样,使用含有60% (v/v)乙腈的缓冲液分离了螺旋霉素、伊维菌素、泰乐霉素、交沙霉素、雷帕霉素和利福霉素。张含智等[67]用CE测定了环肽抗生素硫酸多黏菌素中4种多黏菌素B成分,分离缓冲液为含有HP-β-CD和异丙醇的磷酸三乙醇胺缓冲液(pH 2.5)。Wingert等[68]用微乳电动色谱建立定量分析抗凝血药利伐沙班的方法,并评价其稳定性。
在灵敏度和精密度不如药物分析常用的HPLC时,CE可通过多种分离模式提供独特的选择性,获得与常用HPLC不同的分离效果。在新药审评中普遍关注的杂质研究方面,应用CE可能分离出常规HPLC未分离而漏检的杂质。这个互补技术的概念也提示,寻找HPLC分析的难点、发挥CE的长处,也是CE研究值得注意的方向。
另一方面,CE在分离效率、选择性、快速与经济性的优势仍然存在,产生CE应用研究目前现状的原因与其灵敏度和精密度密切相关,这两方面的进展都可能显著扩展未来CE的应用领域,对CE的应用研究产生深远影响,值得关注。
2 中药与天然药物分析
2.1 CE分离分析方法研究
Liu等[70]使用甲基-乙烯基咪唑(methyl-vinylimidazole)官能团化有机聚合物单体,自制的整体柱,采用毛细管电色谱-质谱联用技术测定了吴茱萸果实中3种生物活性成分吴茱萸胺(evodiamine)、吴茱萸次碱(rutaecarpine)、吴茱萸内酯(limonin)的含量。李晓慧等[71]制备聚甲基丙烯酸丁酯-乙二醇二甲基丙烯酸酯整体SPE材料,与CE联合分离检测桑叶中芦丁、绿原酸、槲皮素等成分。
曾雪等[72]用加入β-CD的CZE方法测定了三黄片中大黄素和大黄酚。Sereia等[73]使用含HP-β-CD的硼酸缓冲液为背景电解质,建立了巴西药用植物卡图巴(Trichiliacatigua)树皮乙酸乙酯部位多种非对映异构多酚成分的CE-UV测定方法。
2.2 检测方法
Zhou等[74]将聚醚醚酮(PEEK)毛细管用于CE-MS,使用高有机溶剂体系分析中药中多组活性生物碱,表现出良好的分离性能,并应用于浙贝母药材中贝母甲素(peimine)和贝母乙素(peiminine)的高灵敏定量分析。Cheng等[75]用自建的基于小离心管流动溶液电喷雾接口的非水CE-MS方法,以二羟基蒽二酮为内标,分离测定了中药大黄中大黄素甲醚、大黄酚和芦荟大黄素。
Sun等[76]采用CE-电致化学发光检测法测定了石蒜中加兰他敏(galanthamine)、高石蒜碱(homolycorine)、石蒜宁碱(lycorenine)和多花水仙碱(tazettine)4种生物碱,检出限依次为14、11、1.8和3.1 ng/mL。孙双姣等[77]还测定了红花石蒜中伪石蒜碱、石蒜碱和高石蒜碱,通过添加聚乙烯吡咯烷酮(PVP)改善了CE的分离效果。邓光辉等[78]以CE-电致化学发光检测法,建立了金钗石斛中石斛碱含量的测定方法,检出限为0.045 mg/L。他们还建立了同时检测博落回果实中血根碱和白屈菜红碱含量的CE电致化学发光分析方法,分离缓冲液中使用了含50%的乙腈[79]。
Wu等[80]利用Triton X-100和SDS组成的混合胶束对姜黄素天然荧光的增敏作用,建立了MEKC-激光诱导荧光检测(LIF)测定姜黄、药用姜黄搽剂、咖喱调味料和人尿液中姜黄素、去甲氧基姜黄素、二去甲氧基姜黄素3种姜黄素类成分的方法。唐梦杰等[81]用CE-C4D测定中药材连翘中齐墩果酸、熊果酸和白桦脂酸,检出限为1.0~1.4 μg/mL。
2.3 中药指纹图谱
CE中药指纹图谱的研究发挥了CE分离复杂样品的优势,这方面的研究文献较多。Hou等[82]提出结合CE指纹谱与线性定量分析法(linear quantitative profiling)评价苦参的质量一致性。张蕾等[83]以硼酸盐-SDS-无水乙醇为分离缓冲液,建立了新疆紫草的MEKC指纹图谱,确定了6个特征峰,其中两个峰被识别为左旋紫草素和乙酰紫草素。孙姗等[84]用CZE建立了苣荬菜的指纹图谱,确认9个共有峰,并识别其中3个峰分别为绿原酸、芦丁和槲皮素-3-β-D-葡萄糖苷。李成思等[85]用MEKC建立有12个共同峰的藏药白花龙胆指纹图谱。郭鹏等[86]采用场放大进样的CE方法测定了中药附子的指纹图谱,所采集的10批药材中有8个共有峰,识别出其中苯甲酰次乌头原碱、苯甲酰新乌头原碱和苯甲酰乌头原碱3个峰。陈宝龙等[87]用MEKC建立了山楂药材的指纹图谱,确定包括金丝桃苷在内的16个共有峰。王玲娇等[88]建立了木香顺气丸CE指纹图谱,以橙皮苷为参照峰,确定了14个共有指纹峰。孙姗等[89]用CE研究了白车轴草的毛细管电泳指纹图谱,确认了10个共有峰。惠阳等[90]建立了中药材高良姜黄酮类提取物的MEKC特征图谱,确定了15个共有峰,并识别其中6个峰分别为儿茶精、山柰酚、槲皮素、高良姜素、山柰素-4′-甲醚和姜黄素。
张政等[91]建立了CE检测全蝎酶解液中蛋白类成分(>10 kDa)的指纹图谱分析方法,并对10批样品做了相似度评价,初步确定了以25个峰为共有峰的指纹图谱。陈莉等[92]建立了中成药康复新液(从美洲大蠊制得)的CE指纹图谱,确定了8个共有峰。
2.4 中药多糖分析
依明·尕哈甫等[93]经α-萘胺-氰基硼氢化钠(NaBH3CN)衍生,用CE测定了牛舌草多糖水解后的单糖组成。张建等[94]以1-苯基-3-甲基-5-吡唑啉酮(PMP)为衍生试剂,用CZE测定不同虫草菌粉制剂中虫草粗多糖的单糖组成。
2.5 中药分析其他方面
Alzoman等[95]建立了CE测定三花六道木(Abeliatriflora)植物叶子粗提物中黄芩苷和咖啡酸的方法。Tascon等[96]建立了CE同时测定常见植物中去氢骆驼蓬碱(harmine)、骆驼蓬碱(harmaline)、哈尔酚(harmol)、去甲骆驼蓬碱(harmalol)、去甲氧去氢骆驼蓬碱(harmane)和去甲哈尔满(norharmane)6种具有精神活性的β-carboline类生物碱的方法,用该方法分析骆驼蓬种子输液,检出了前3种成分。杨霞等[97]用CE同时检测决明子中大黄素、大黄酸、大黄酚、大黄素甲大黄素甲醚和橙黄决明素5种蒽醌类化合物。王博等[98]用MEKC同时测定了小青龙颗粒中麻黄碱、伪麻黄碱、甲基麻黄碱与芍药苷的含量。
卢恒等[99]还建立了人参中铜和镉离子的MEKC-LIF测定方法。样品用浓硝酸-过氧化氢彻底消解后,用荧光络合剂异硫氰酸荧光素酯-偶氮乙二胺四乙酸柱前络合其中的Cu2+与Cd2+,以pH 9.3的硼酸盐-SDS为分离缓冲液分离,激发波长为483 nm,在520 nm波长下荧光检测。
中药作为复杂样品,为CE的研究提供了广阔的舞台。目前需要考虑CE是否可以深入研究中药这类复杂样品在HPLC分析中遇到的难题。其中,CE-MS分析中药的研究值得关注。
3 体内药物分析
Kubáň等[100]综述了CE用于非传统体液样品(母乳、呼吸冷凝物、汗液、唾液、羊水、脑脊液或干血斑)中1 000 Da以下小分子(离子)成分的分析。Silva等[101]综述了测定体液样品中摇头丸成分的分析方法。
3.1 新方法研究
Opekar等[102]提出一种可从汉密尔顿(Hamilton)进样器进样分析μL级临床样品的CE分析自动微进样器。注射器针头的出口与分离毛细管的进样口对准并相距数百μm,进样时从注射器中挤出的一滴样品在毛细管的入口被捕获进入到毛细管,洗掉多余的样品溶液后采用CE分离。方法测定总量10 μL鼠血浆中的抗寄生虫药戊烷脒(pentamidine),使用C4D检测器的定量限是8 μmol/L。Emara等[103]建立了毛细管柱内衍生化结合荧光检测的快速MEKC测定人血清中吗啡的方法。在含有铁氰化钾的背景电解质中,吗啡被氧化成高荧光产物伪吗啡,方法检出限为1.14 ng/mL,并用于单次口服硫酸吗啡控释片后血清中吗啡的测定。Wu等[104]将荧光染料7-(二乙胺基)香豆素-3-羧酸作为衍生化试剂用于405 nm半导体LIF检测,氢氯噻嗪、氯噻嗪、氯噻酮衍生后经MEKC基线分离后的检出限分别为0.24、0.29、和0.23 nmol/L,用于加标人尿液样品的分析。
3.2 体内药物分析的检测灵敏度
CE-MS和CE在线浓缩的方法得到较多应用。Piestansky等[105]研究和比较了两种用于监测人尿中戒烟药伐尼克兰(varenicline)的CE方法。一种是CZE-MS快速分析方法(分析时间7 min),另一种是低成本的基于在线样品浓缩的CZE-UV方法,两法都做了方法验证。
3.2.1CE-MS
Hernández-Mesa等[106]用CE-MS/MS建立了经分子印迹固相萃取处理的尿液样品中11种5-硝基咪唑类化合物及其代谢物的分析方法,检出限为9.6~130.2 μg/L。vidrnoch等[107]用非水CE-MS/MS建立了同时测定人血清中9种苯二氮卓类药物(bentazepam、etizolam、deschloroetizolam、diclazepam、flubromazepam、flubromazolam、nimetazepam、phenazepam、pyrazolam)的方法。采用25 mmol/L醋酸铵和100 mmol/L三氟乙酸的乙腈溶液为分离电解质,测定的LOD在1.5~15.0 ng/mL之间。Tejada-Casado等[108]用MEKC-MS/MS建立了同时测定动物尿液中13种苯并咪唑的新方法。使用在线扫集模式改善灵敏度,LOD低于70 μg/L。
Sun等[109]将构建的新型电致化学发光传感器与毛细管电泳联用,测定了人血浆中盐酸喹那普利(quinapril)及其代谢物盐酸喹普利拉(quinaprilat), LOD分别为3.6 ng/mL和3.9 ng/mL。李享等[110]基于碱性溶液中待测组分对[Ag(HIO6)2]5-(二(过碘酸氢)合银(Ⅲ)配离子)-鲁米诺体系化学发光的抑制作用,建立了测定人血清中万古霉素和去甲万古霉素的SPE-CE-间接化学发光检测方法,检出限均为2.5 μg/mL。该团队的朱怀娇等[111]则利用吗啡对相同体系化学发光的抑制作用,建立了检测尿液与血液中吗啡含量的CE-间接化学发光检测方法,检出限为0.75 mg/L。Saar-Reismaa等[112]用CE荧光检测快速分析3种致幻成分,包括唾液样本收集过程,可在大约15 min内完成摇头丸滥用的检测。
3.2.2在线浓缩
3.3 其他体内药物分析研究
李兴华等[121]建立了高效毛细管电泳-紫外检测法快速分离测定10-羟基喜树碱、6-巯基嘌呤、5-氟尿嘧啶3种抗肿瘤药物的方法。刘晓凤等[33]用CE测定给药大鼠血浆中头孢地尼浓度,并计算了药动学参数,定量限为0.2 μg/mL。胡小波等[122]建立了血浆直接进样测定大鼠血浆中丙吡胺游离浓度和总浓度的CE方法。Liu等[123]建立简便的CZE方法,在自杀基因抗肿瘤疗法中,可同时检测携带CD/5-FC自杀基因系统的人细胞中5-氟胞嘧啶及其目标转化产物5-氟尿嘧啶。有关手性药物体内分析已在1.7节提及。
体内药物分析对检测灵敏度有较高要求,LC-MS/MS具有优势地位。CE的样品使用量少,通过在线浓缩与CE-MS/MS建立起新的技术特色,在体内药物分析领域有较好的发展潜力。
4 生物制品药物分析
CE已经成为生物药(包括单克隆抗体)完整状态和middle-up途径分析表征的基本工具。Kahle等[124]综述了蛋白质电荷异质性分析的3种CE技术:cIEF、全柱成像cIEF(icIEF)和CZE,讨论了各方法实验参数对分析结果影响的研究进展,总结了各方法精密度的进展,提出开发cIEF分析方法时的参数和范围,对相关实验具有较好的实用价值。陈泓序等[125]从纯度分析、等电点(pI)测定、电荷异质性分析和N-寡糖分析几方面详细综述了CE在单克隆抗体药物分析中的应用。高凡等[126]综述了MEKC与MEKC-MS在蛋白质分离分析方面的研究进展。
4.1 CE-SDS
《中国药典》2015版中收录的“单抗分子大小变异体测定法”(通则3127)是目前单克隆抗体纯度检测的主要方法,使用含有十二烷基硫酸钠和亲水性聚合物的分离缓冲液,称为CE-SDS。Gu等[127]使用计算机模拟和CE-SDS实验,评估了基线干扰对治疗性蛋白药物纯度测定的影响,认为基线干扰源自样品分析中的热干扰以及背景电解质的基线特征。他们还提供了改善准确度的实验建议。
Zhang等[128]采用SDS-毛细管凝胶电泳(CGE)和柱头场放大样品堆积在线预浓缩技术,仅用25 ng总蛋白实现了基于分子大小的蛋白质分析。在214 nm下,病毒外壳蛋白的检出限为0.2 ng/mL (3.3 pmol/L),灵敏度增强3个数量级,可与银染SDS-聚丙烯酰胺凝胶电泳(PAGE)相媲美。方法已用于分析不同血清型和转基因的腺相关病毒(AAV)产品的纯度。
Beckman等[129]使用CE-SDS分析RTP-1(一种Fc-Adnectin融合蛋白,约80 kDa)时,分离度和峰对称性不理想,将SDS换成十六烷基硫酸钠(sodium hexadecyl sulfate, SHS)后,其分离度和塔板数分别提高了2.3倍和8倍(见图3)。

图 3 凝胶缓冲溶液中添加和不添加0.2% SHS的RTP-1蛋白CGE电泳图[129]Fig. 3 CGE electropherograms of RTP-1 comparing results with or without 0.2% SHS added to the gel buffer solution[129] CGE: capillary gel electrophoresis ; SHS: sodium hexadecyl sulfate; SDS: sodium dodecyl sulfate.
Kahle等[130]使用不同相对分子质量的蛋白质混合物比较了主要的商品化CE仪器用于CE-SDS蛋白分析的性能,有助于用户根据实际需求选购。
刘振东等[131]研究优化了分析非还原单克隆抗体药物纯度的前处理条件,认为高pH的样品缓冲液中封闭剂降解,易使未封闭的游离巯基介导链间二硫键断裂,会导致样品处理过程中出现非预期降解。邓钦培等[132]用CE-SDS建立了测定人绒促性素(hCG)解离亚基的测定方法,并用于工艺开发及稳定性研究中检测影响其活性的亚基解离现象。
4.2 生物制品的CE-MS研究
CE-MS联用是生物药物分析的热点。Sanchez-Hernandez等[133]提出在CE-SDS与MS联用分析抗体时在线消除SDS干扰的方法。通过在样品进样区带后注入阳离子表面活性剂的甲醇-水溶液,利用阳离子表面活性剂与SDS在管内相对移动和结合,除去与抗体结合的SDS,使抗体的CE峰变窄变强。例如含有0.2% (v/v) SDS的样品中,使用十六烷基三甲基溴化铵或苯扎氯铵分别可恢复97%和95%的质谱峰强度。该方法也被用于其他蛋白质和溶于10%SDS中的抗体,以及其他包含SDS的基质。
Le-Minh等[134]提出在天然活性条件下的CE-MS方法,在没有去活和去折叠的活性条件下分析完整的治疗药物英利昔单抗。非去活的实验条件保护了单抗的构象差异和自缔合,CE毛细管使用了聚凝胺(polybrene)-硫酸葡聚糖-聚凝胺3层涂层,鞘流液为异丙醇-水-醋酸,单次分析可以同时检测天然的和去折叠的单体以及二聚体。
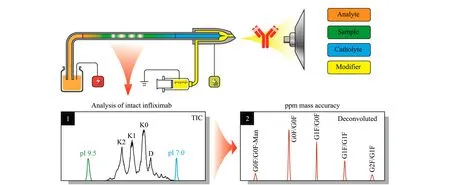
图 4 用CIEF-MS方法分析英夫利昔单抗的变体[135]Fig. 4 Identified variants of infliximab by cIEF-MS[135]cIEF: capillary isoelectric focusing.
Wang等[135]用3处通孔的小塑料离心管构建流动溶液接口建立了高分辨cIEF-MS联用方法,不用抗对流剂甘油时pI的分辨率可以达到0.02 pH。以英夫利昔单抗制剂为样品,用轨道阱(Orbitrap)质谱成功分离了4个变体,并在10-6质量精度下识别出13个英夫利昔单抗分子量变体(见图4)。该研究团队的Cheng等[136]使用类似的此前建立的非水CE-MS联用接口,以含20 mmol/L甲酸铵的乙腈-甲醇-甲酸(20∶78∶2, v/v/v)为分离缓冲液、含2 mmol/L甲酸铵的乙醇溶液为接口溶液,分离了6种高疏水性抗菌肽temporin样品。该团队[75]还将此非水CE-MS接口用于中药成分。
Haselberg等[137]用低流量无鞘CE-MS在完整和middle-up策略(使用内切蛋白酶IdeS)分析完整和酶切后的单克隆抗体。通过分析两种单价纳米抗体Nb1和Nb2(各自包含Myc-6xHis标记)以及两种Nb1结构域的二价抗体(Nb1-35 GS-Nb1)对系统进行评价,并用于分析几种具有不同电荷和糖基化差异的单抗药物。
Camperi等[138]对人绒毛膜促性腺激素(hCG)进行了完整水平的CE表征,CE分离两种hCG生物类似药物:Ovitrelle(重组r-hCG)和Pregnyl(分离自孕妇尿液),结果两者峰数、迁移时间和峰强度均不相同,说明不同来源的hCG药物中含有不同性质和比例的亚型。经cIEF分析,两者pI范围分别为3.4~4.7和4.5~5.2。他们进一步开发出CZE方法,可以区分对应不同的hCG亚型的至少6个峰,建立了可用于hCG各种异构体指纹识别的CZE-MS联用分析方法。
4.3 全柱成像毛细管等电聚焦法
icIEF在检测多肽与蛋白质药物的等电点以及电荷异质性方面有明显优势,应用较多。Goyon等[139]用icIEF测定了23个美国食品和药品管理局(FDA)和欧洲药物管理局(EMA)批准的pI范围从6.1到9.4的单克隆抗体治疗药物,并建议icIEF可以作为此类测定的参考技术。Demirdirek等[140]报道了监测静脉输液中葡萄糖参与的蛋白糖化产物的icIEF方法,显示其能够检测不稳定的席夫碱糖基加合物。
武刚等[141]考察了icIEF测定单抗等电点的实验条件(包括添加尿素、聚焦时间、pI标记物)对icIEF测定抗HER2单抗、抗VEGF单抗的主峰等电点测定的影响,认为pI标记物的选择对等电点测定结果影响较大,测得等电点受到pI标记物标注等电点准确性的影响。他们[142]还在icIEF分析单抗电荷异质性时组织国内多家质控实验室对该方法进行验证,并制备出用于icIEF的系统适用性对照品。
李响等[143]用icIEF有效分离了IL-15融和蛋白的12个电荷异构体,并证明唾液酸化程度差异是电荷异质性产生的主要原因。他们[144]还比较了icIEF与CZE两种分析重组人促红素电荷异构体含量的方法,认为两者均可满足rhEPO电荷异构体的质控需求。孟晓光等[145]用icIEF检测多肽与蛋白质药物的等电点,人血红蛋白的4种主要电荷异构体实现了基线分离,评价了进口及国产贝伐单抗一致性。周朝明等[146]用国产icIEF仪器测定了多肽KR-1、胰岛素、融合蛋白疫苗重组人乳头瘤病毒的等电点。
4.4 抗体药物分析
Ladner等[147]将单抗的胰蛋白酶消化和电泳分离水解物在线自动化联用,用于3种单克隆抗体(曲妥珠单抗、英夫利昔单抗和托西珠单抗)的分析,结果与离线消化的谱图相当,可用于单克隆抗体药物的质量控制。Said等[148]利用无鞘CE-MS/MS,从整体、middle-up和bottom-up不同的分析策略对抗体偶联药物本妥昔单抗(brentuximab vedotin)的结构进行表征。Hutanu等[149]使用盐酸胍溶液冲洗以避免管壁蛋白质的吸附,通过加入特异性配体,利用部分填充毛细管的亲和CE法分离了两种相似单克隆抗体的混合物。
王文波等[150]用CZE建立IgG2型单抗电荷异质性分析方法时,使用含有羟丙基甲基纤维素的6-氨基己酸-三乙基四胺缓冲液作为分离缓冲液,改善了样品的分离。黄碧君等[151]用CE测定重组人源化IgG2抗体的纯度,使用的是商品化的SDS-MW Analysis Kit分离缓冲液。
4.5 N-寡糖分析
CE已经具有分离相同单糖序列但位置不同的寡糖同分异构体,并可区分单糖之间的α连接和β连接。Lu等[152]详细综述了2014~2018年初CE分离N-寡糖的进展和主要应用。Szigeti等[153]提出用温度梯度技术改进荧光标记N-寡糖CE分离的选择性。在15~45 ℃的温度区间内以5 ℃为间隔梯度升温,分离了3种寡糖的混合物,显示温度是CE寡糖分离的重要实验参数。李凤等[154]以8-氨基芘-1,3,6-三磺酸钠(APTS)为荧光衍生试剂,分别建立中药多糖中单糖组成的CE分析方法,以及人血清中糖蛋白N-寡糖的CE指纹谱分析方法。Savicheva等[155]还通过对APTS的结构改造,得到用于还原糖分析、具有更高荧光强度和更多负电荷的CE激光诱导荧光衍生试剂。
4.6 二硫键分析
表征二硫键的连接位置仍然是分析肽/蛋白质药物的一个重要问题。Delvaux等[156]结合CZE-MS、离子迁移谱-MS和理论计算,研究了具有两个分子内二硫键,但存在不同半胱氨酸连接方式的3种多肽的二硫键异构体,在水相(CZE)和气相(离子迁移谱)中的分离。
4.7 生物制品分析的其他方面
Liang等[157]提出一种用于改善CZE分离高浓度组分的扩展速度间隙技术(stretching velocity gap, S-VGCE),其机理是高浓度的样品成分先被拉伸成较宽的区带,再通过电切换的切割效应将样品分成多个短区带,然后将这些短区带分离开。Liang等[157]将该技术用于分离含有溶菌酶、牛血清白蛋白和核糖核酸酶组成的混合蛋白质样品。Maldaner等[158]以雷尼替丁为内标,用CZE分析了重组人甲状旁腺素(rhPTH 1-34),线性范围为0.25~250 μg/mL。Kpaibe等[159]报道了蛇毒液指纹图谱的CZE分析方法,使用改性涂层毛细管以避免蛇毒中大量蛋白质和肽的吸附,可用于不同批次蛇毒液的质量控制。Yao等[160]建立了CZE分离测定门冬酰胺酶(埃希)及其酸性变体的方法,定量限为1.9%。
van Tricht等[161]用CE直接定量检测上下游加工样品中完整的腺病毒颗粒,用于开发基于腺病毒疫苗期间的分析监测。使用由125 mmol/L Tris、338 mmol/L三甲基甘氨酸和0.2%(v/v)聚山梨酯-20组成的缓冲液(pH 7.7),将腺病毒颗粒从复杂的混合物中分离出来。生产过程中5个具有代表性的样品中腺病毒的定量测定结果为0.5×1011~1.5×1011腺病毒颗粒/mL (约80~250 pmol/L)。
中国仓鼠卵巢(CHO)细胞是常用于单抗生产的蛋白表达系统,Wang等[162]建立了同时检测CHO细胞中7种氧化还原和能量相关代谢物的毛细管电泳方法。采用在线聚焦技术,7种代谢物的检出限为0.050~0.688 mg/L,并用于两种不同培养条件下CHO细胞提取物的分析。
Hu等[163]报道了一种利用毛细管电泳一体化固定化酶反应器在线检测青霉素酶活性和抑制作用的方法,可用于细菌耐药性的治疗和诊断。Chen等[164]以青霉素G为底物,建立了CE分析丝氨酸-β-内酰胺酶和金属-β-内酰胺酶的方法,并对在线和离线两种方法都做了方法认证。
吴克等[165]用CE紫外间接检测法测定了四价轮状病毒疫苗原液中蔗糖的含量,分离缓冲液为含CTAB的2,6-吡啶二羧酸-NaOH溶液(pH 12.6)。李响等[166]用CZE测定了重组人干扰素α-1b滴眼液中间甲酚的含量。
在生物制品分析方面,CE-SDS、icIEF已经形成了方法优势并成为蛋白类药物法定分析方法难以替代的一部分,CE-MS也具备了在生物制品分析中解决实际问题的能力。生物制品在新药研发和生产过程中提出了很多需要解决的分析问题,未来在这方面CE将会得到越来越多的应用。
5 结论
CE在药物分析方面仍然具有特色和优势,并显示出较大的发展空间。CE-MS在不断改进接口技术的同时,在药物分析各方面都得到广泛应用,MS和MS/MS强大的定性能力与优越的检测灵敏度弥补了CE的短板,而CE对包括生物大分子在内的强极性生物活性成分的分离能力也使CE-MS显示出独特的优势。用于蛋白类药物分析的icIEF和CE-SDS在应用中逐渐走向成熟,成为生物制品分析的有力工具,在作为国内外研发热点的抗体药物研究方面更具优势,两者均已经实现与MS联用,并会显示出更多应用潜力。广泛应用的在线样品浓缩技术使检测灵敏度不再成为CE分析的难题。对分析重复性的改进以及便携式仪器的出现可能使国内CE仪器拥有走向世界的技术特色,也为CE更多进入日常药品检测实验室以及快检现场提供了想象空间。
这些研究成果显示,CE分析的多个重要瓶颈问题(例如灵敏度、重复性、定性能力等)已经出现突破的迹象,令人期待。相对于其他色谱分析方法,CE技术的掌握还普遍存在一定难度,将这些成果和方法用于解决药物分析中存在的很多具体问题还需要各方面的耐心和投入。在应用研究过程中不断产生的新问题和新思路,以及CE在方法学和其他领域研究成果的应用,推进着CE药物分析的不断发展。
致谢 感谢上海市食品药品检验所季申教授在中药分析方面的讨论。
