金属有机骨架101(Cr)-固相萃取环境水体中分散染料
2020-09-17高仕谦李小蒙鲍秀敏景建军邵子纯王思远赵德泽
高仕谦, 李小蒙, 鲍秀敏, 景建军, 邵子纯, 王思远, 赵德泽
(1.苏州科技大学环境科学与工程学院,江苏苏州 215009;2.苏州国环环境检测有限公司,江苏苏州 215011)
偶氮类分散染料含有重氮基(-N=N-),空间排序为立体基阵式。蒽醌类染料4个α位上引入的供电子基团(-OH、-NH2)可提供良好的取代基体[1],以上二者稳定的结构特性,使它们的水溶性较差,在长时间的光照和生物作用下降解效率极低[2,3]。该类染料进入人体后,可被代谢分解为芳香胺类致癌物质而残留于体内,激活细胞内的原癌基因,进而导致高致癌的风险[4]。目前,一些国家或国际组织严禁该类染料印染制品进入市场[5]。当这类染料进入环境水体中,可能会危害到地下水资源和供给水源地的安全,进而威胁到人们的健康安全[6]。因此,建立一种高效检测该类染料的技术具有现实意义。
由于同类分散染料的结构及化学性质较为相似,使得常规高效液相色谱(HPLC)在分散染料多组分分析过程中不能获得良好的分离效果[7]。因此可以采用超高效液相色谱与质谱联用技术对产生的相似质谱裂解碎片进行分析,以实现对多种染料的同步定性定量分析[8,9]。现有文献报道的样品预处理方法主要有聚酰胺粉吸附法[10]、超声辅助萃取法[11]、固相萃取法[12]以及分散固相萃取法等。采用分散固相萃取对水样进行前处理,具有具有快速、简便、高效等优点等优点[13]。
MOF-101(Cr)属于金属有机骨架纳米材料,其是由含碳、氮、氧等多齿有机配体与金属离子Cr(Ⅲ)自组装而成。该材料含有两个直径分别为34 Å和29 Å的孔笼结构[14],使其比表面积较大、孔隙率较高,拥有大量的不饱和的金属位点,同时化学性质比较稳定,已在气体储存、工业催化以及有机污染物分离等方面得到广泛应用[15 - 17]。分散染料结构中富含苯环及杂原子p-π共轭体系,可与MOF-101(Cr)中的六元碳环发生静电作用而被吸附。本实验以MOF-101(Cr)为吸附剂构建分散固相萃取体系,建立一种环境温和、高效且回收率良好的前处理方法,结合超高效液相色谱-串联质谱法(UPLC-MS/MS)s,实现了对实际水体中分散蓝35A、分散红1、分散蓝106、分散橙37和分散棕1共5种分散染料的定量分析。
1 实验部分
1.1 主要仪器及试剂
TSQ Quantum Ultra EMR三重四极杆质谱仪、Ultimate 3000超高效液相色谱仪(美国,Thermo公司);VD115真空干燥箱(德国,Binder公司);SK-1快速混匀器(金坛市科析仪器有限公司)。
分散蓝35A(95.0%)、分散红1(95.0%)、分散蓝106(100.0%)、分散橙37(99.0%)和分散棕1(50.3%),均购自德国Dr.Ehrenstorfer公司;Cr(NO3)3·9H2O(99.0%)、2-氨基对苯二甲酸(98.0%)、对苯二甲酸(98.0%)、NaAc(99.9%)和甲酸(98.0%),均购自上海阿拉丁有限公司;无水乙醇、丙酮、二氯甲烷、乙酸乙酯和氨水均为分析纯,购自上海国药。乙腈、甲醇和冰HAc均为色谱纯(Tedia)。水为去离子水。

图1 MOF-101(Cr)的化学结构Fig.1 The chemical structure of MOF-101(Cr)
1.2 材料的制备
Cr(NO3)3·9H2O与对苯二甲酸按质量比2∶1混合后,加入至0.05 mol/L NaAc溶液中,于高压反应釜内200 ℃下反应12 h,缓慢冷却至室温,过滤,滤饼经无水乙醇和水交替洗涤3次后,置于反应釜中,用无水乙醇于90 ℃下纯化6 h,冷却至室温后过滤。滤饼于90 ℃下真空干燥12 h,得绿色粉末产品,即为MOF-101(Cr)纳米材料,化学结构见图1。
1.3 分散固相萃取过程
准确称量8.0 mg MOF-101(Cr)于含有2.0 μg/L目标物的10 mL水样中,调节pH=6.0,振荡吸附7 min,4 000 r/min 离心,弃去上清液得沉淀物。加入0.5 mL乙醇∶二氯甲烷=1∶2(V/V),振荡解吸0.5 min,离心得洗脱液,该过程重复3次,合并洗脱液,N2挥至近干,初始流动相回溶至1.0 mL,经0.22 μm有机滤膜过滤后,进行UPLC-MS/MS分析。
1.4 色谱和质谱条件
色谱柱为Zorbax Eclipse XDB-CN柱(150 mm×4.6 mm,3.5 μm);流动相为0.1%甲酸水溶液(A)和乙腈(B),采用等度洗脱:20%A∶80%B;流速为0.5 mL/min;柱温为30 ℃;进样量为10 μL。电喷雾离子源为正离子(ESI+)模式;喷雾电压为3.5 kV;喷雾温度为300 ℃;鞘气压力为25 MPa;辅助气压力为10 MPa;离子传输管温度为350 ℃。色谱及质谱分析参数如表1所示。

表1 5种分散染料的色谱保留时间和质谱参数
2 结果与讨论
2.1 材料表征
采用扫描电镜(SEM)和透射电镜(TEM)对MOF-101(Cr)进行形貌分析。如图2(a)、2(b)所示,制备所得MOF-101(Cr)纳米材料具有典型的正八面体晶体结构、形貌均匀、粒径尺寸均一且紧密堆叠。
MOF-101(Cr)的X射线衍射(XRD)如图2(c)显示,MOF-101(Cr)的特征峰与文献报道[18]一致,表明MOF-101(Cr)已成功制备。如图2(d)所示,该复合材料的比表面积、平均孔径和孔隙体积分别为302.3 m2/g、5.10 nm和0.15 cm3/g。氮吸附-解吸等温线呈Ⅳ型,在极高蒸气压下出现迟滞现象,表明MOF-101(Cr)有介孔特性,证明其在吸附过程中易发生毛细凝聚现象,使其表面的吸附量急剧增加。
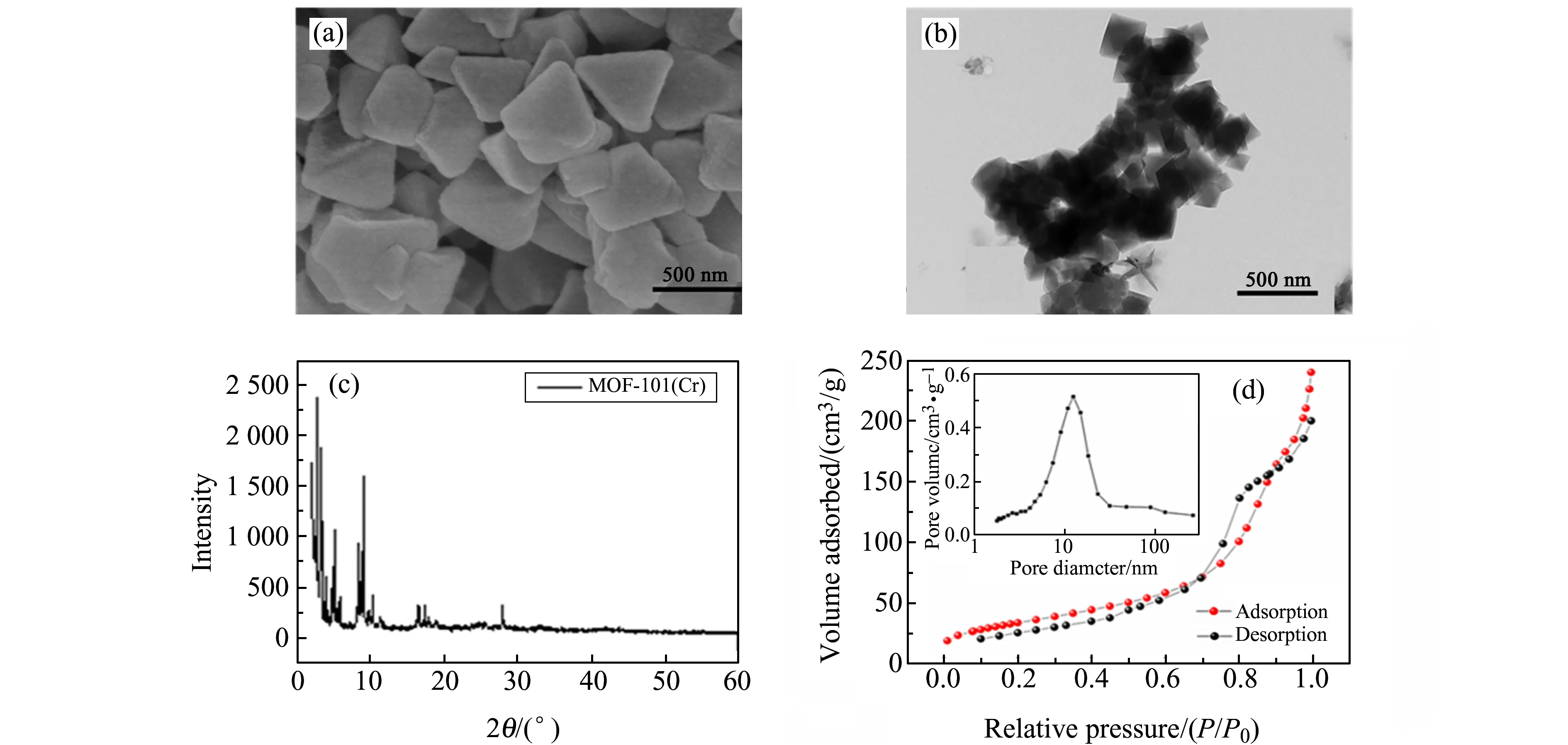
图2 MOF-101(Cr)的SEM(a)、TEM(b)、XRD(c)和BET/BJH(d)图Fig.2 SEM(a),TEM(b),XRD(c) and BET/BJH(d) images of MOF-101(Cr)
2.2 固相萃取条件选择
2.2.1 MOF-101(Cr)用量分散染料具备接受质子和提供电子对的能力,具有一定的碱性和亲核特性,而MOF-101(Cr)配体中心原子Cr(Ⅲ)可提供亲核空轨道;同时,染料中的苯环与MOF的苯环可形成π-π共轭,进而实现对目标污染物的吸附。考察MOF-101(Cr)的用量(4.0~12.0 mg)对分散染料萃取效率的影响(图3a)。当MOF-101的用量从4.0 mg增加到8.0 mg时,分散性染料的回收率相应提高,并在8.0 mg时,回收率达到最大值,范围是59.8%~68.9%;当继续增加MOF-101(Cr)的用量时,回收率基本平衡。因此,实验中选择8.0 mg为最佳吸附剂用量。
2.2.2 固相萃取pH值MOF-101(Cr)的等电点为9.6,当pH<9.6时,MOF-101的表面为正电荷[19],非离子型染料部分溶于水中主要是依靠其含有的亲水基团-NH2和-OH。溶液pH值的变化会改变吸附剂表面的Zeta电位和目标污染物对水的亲和力,从而影响MOF-101(Cr)与分散染料的分子间静电作用力的强弱,进而影响富集效率。考察水溶液pH值为3.0~9.0时,对分散染料的富集效率的影响。如图3b所示,当pH=6.0时,MOF-101(Cr)的萃取效率最高,在碱性溶液中的平均富集效率明显优于酸性溶液。因此,本实验最佳pH为6.0。

图3 MOF-101的用量(a)和溶液pH(b)对回收效率的影响Fig.3 Effects of MOF-101 amount (a) and pH (b) on the recovery
2.2.3 洗脱过程的优化考察有机溶剂对目标物洗脱效率的影响,如甲醇、乙腈、丙酮、无水乙醇、乙酸乙酯和二氯甲烷。如图4a所示,乙醇和二氯甲烷的洗脱能力优于乙腈、丙酮和乙酸乙酯。而采用甲醇洗脱时,大量MOF-101(Cr)悬浮于溶液表面,洗脱效果相对较差。因此本选择乙醇和二氯乙烷组成的二元洗脱体系作为洗脱剂,考察其对洗脱效率的影响。比例分别为1∶3、1∶2、1∶1、2∶1、3∶1,结果如图4b所示。当乙醇∶二氯甲烷组成比例为1∶2时的回收率为67.1%~75.1%。主要是由于分散染料分子结构中含有非离子极性基团,如-OH、-NH2、-CN和-CONHR等,二元洗脱溶剂的协同作用使洗脱溶剂与分散染料间的相互作用力增强,有助于提高洗脱剂的选择性。因此选择1∶2作为适宜洗脱剂类型。
优化洗脱剂用量、洗脱次数对洗脱效率的影响,结果如图4c显示。洗脱剂用量分别为0.5 mL×3、1.0 mL×3、1.5 mL×3,研究发现,当洗脱剂用量为0.5 mL、洗脱3次时回收率比较理想。
考察单次洗脱时间(0.3~2 min)对洗脱效率的影响,如图4d所示。当0.5 min时,5种分散染料的回收率达到最大值;当洗脱时间继续增加时,除分散蓝35A和分散棕1的回收率略有较低外,其他均保持恒定。综合考虑,选择0.5 min作为单次洗脱时间。
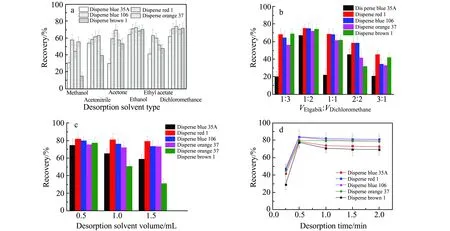
图4 洗脱过程对回收率的影响Fig.4 Effect of eluent process on the recovery

图5 吸附时间对回收率的影响Fig.5 Effect of extraction time on the recovery
2.2.4 吸附时间的优化本实验考察了吸附时间为1、3、5、7、9 min的影响,如图5所示。5种分散染料的回收率均在7 min时达到80.0%以上,而随着吸附时间逐渐增加,回收率基本稳定,因此选择7 min为最佳吸附时间。
2.3 工作曲线、线性范围和检出限
混合标准溶液浓度为0.1~100 μg/L,采用上述色谱和质谱条件进行测定,以标准溶液浓度为x及峰面积为y建立线性关系并绘制标准曲线,测试结果如表2所示。5种分散染料的相关系数(r2)在0.9950~0.9990之间,分散染料检出限(S/N=3)均低于0.037 μg/L。日间精密度(RSD)为5.5%~8.4%(n=3)。数据显示方法有优良的精密度和再现性。
2.4 实际水样的测定
待测水样取自本市3家印染厂出水口,经过0.22 μm有机滤膜过滤,对空白和加标样品进行分析。空白水样1号和3号未检出,空白2号水样品中检测出分散蓝106和分散棕1,浓度分别为0.24 μg/L、0.17 μg/L。加标样品回收率与相对标准偏差如表3所示,结果显示5种染料的平均加标回收率在70.6%~93.5%之间,RSD低于9.4%。因此,方法可被应用于实际污水的检测。
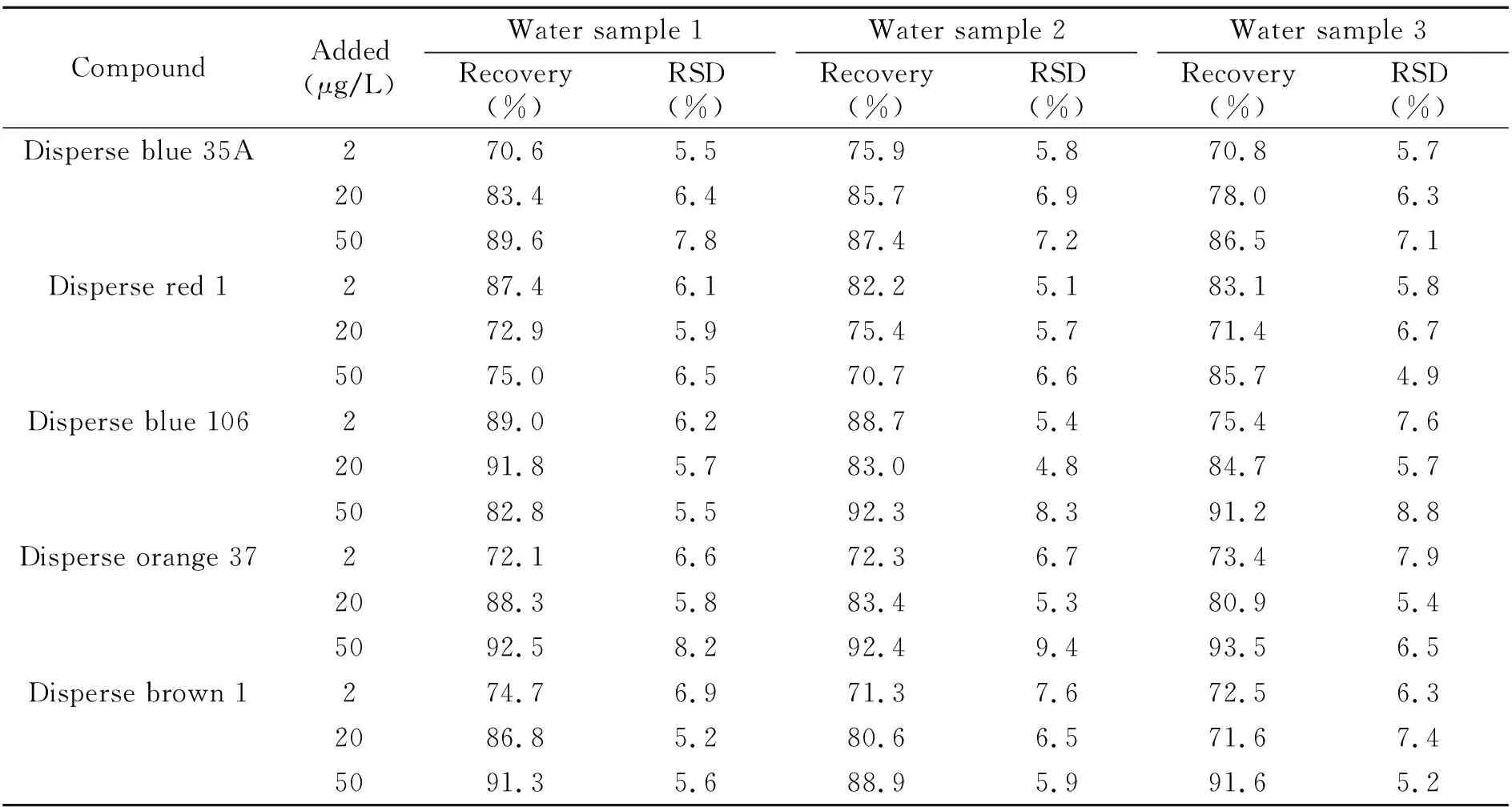
表3 实际水样中5种分散染料的加标回收率(n=5)
将本方法与其他方法进行比较,结果如表4所示。数据表明,本方法引入少量的吸附材料可获得较理想的检出限范围。
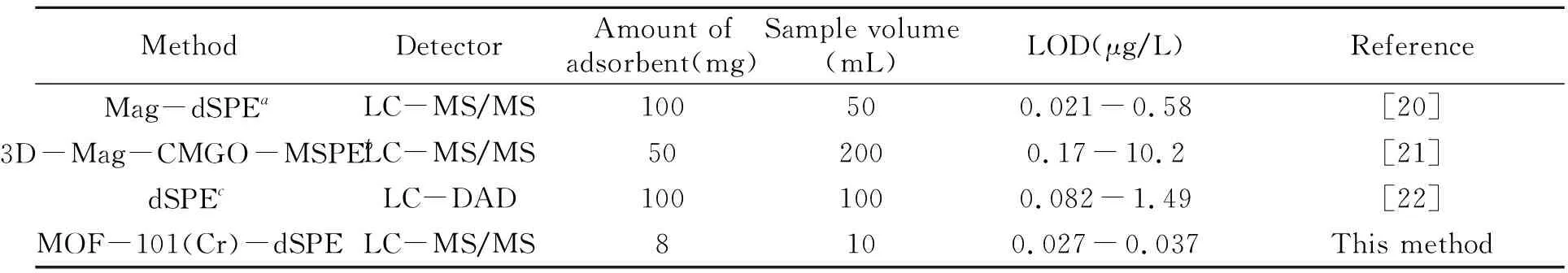
表4 本方法与其他方法对分散染料萃取的比较
3 结论
本研究以合成的MOF-101(Cr)纳米材料作为吸附剂,采用分散固相萃取方法实现对水体中的5种分散染料的富集,结合超高效液相色谱-串联质谱法对其进行定量分析,并且实现对印染厂出水口的实际水样进行检测。方法加标回收率在70.6%~93.5%之间,相对标准偏差低于9.4%。结果表明该吸附剂适用于富集实际水体中的痕量分散染料目标污染物的富集分析。
