类固相法快速合成小颗粒方沸石的研究
2020-09-15郭宏飞胡忠宽刘秀伍谭朝阳曹吉林
张 恕, 赵 斌, 杨 凯, 郭宏飞, 胡忠宽, 刘秀伍, 谭朝阳, 曹吉林
(河北省绿色化工与高效节能重点实验室, 河北工业大学 化工学院, 天津 300130)
1 前 言
方沸石(Na[A1Si2O6]·H2O)作为一种由4,6,8 元环形成不规则孔道的微气孔沸石[1],具有水热稳定性好、选择性高、离子交换性好等优良性能[2-3],被广泛应用于气相净化与分离、土壤修复、水污染处理等领域[4]。其中,天然方沸石主要应用于含氟离子[5]、碳酸根离子[6]以及重金属离子[7-8]等废水处理和放射性元素的吸附[9];由于天然方沸石储量少、纯度低、粒度大等方面无法满足工业生产需求,在环境污染节能控制、烃分离和双功能催化等应用领域需要采用合成方沸石[10]。小颗粒分子筛具有孔道短、活性点位分布广泛等特点,在炼油和石油化工等反应中表现出优异的催化性能[11]。由此可见,小颗粒的合成方沸石具有较高的应用价值和良好的市场前景。
目前众多国内外学者采用多种工艺方法以多种不同原料合成组成和性能各异的方沸石,其中水热合成法应用最为广泛,该法具有操作简单、产品晶型规整等优点。蔡坤川等[12]以铝粉和二氧化硅为原料采用水热合成方法成功地合成了方沸石产品,结果表明调整钠铝硅比为3:1:3,晶化温度高于180 ℃,晶化时间大于12 h,均可合成粒径为5~20 μm 的高纯度方沸石产品。王国栋等[2]以粉煤灰为原料,水热法合成了方沸石,结果表明按一定合成比配料,190 ℃水热晶化12~24 h,均能合成较纯净的方沸石产品,产品孔容积为0.028 cm3·g-1,表观主孔径约为3 nm,等效比表面积为11.194 m2·g-1。刘泽等[4]以钢渣、粉煤灰为原料,按照n(钢渣):n(粉煤灰)=4:6,n(SiO2):n(Al2O3)=4:2 配料,在晶化温度为150~180 ℃下水热晶化12 h,可以合成纯度较高、晶相较好、热稳定性较高的方沸石。WIERSEMA 等[13]以纯硅为硅源,调节初始凝胶组成为Al2O3:84SiO2:87Na2O:2560H2O,在130~160 ℃晶化一定时间合成了方沸石产品,该研究证明了方沸石晶体生长速度受晶化温度控制,且硅源杂质在一定程度上可以促进晶核形成过程。
传统水热方法合成方沸石的工艺存在晶化温度高、晶化时间长等诸多弊端,开发绿色合成方沸石的工艺路线、优化传统工艺条件成为当前方沸石研究的焦点问题。TATLIER 等[14]分别在120 和180 ℃下,对微波辐射和常规加热法合成方沸石的过程进行了研究,结果表明微波辐射加热可以提高方沸石的结晶速率,极大地缩短了合成周期,但此法操作复杂,难以实现工业化生产。WANG 等[15]利用C18-2-8Br2作为双向表面活性剂,采用双功能模板一步法,在140 ℃下水热晶化96 h 成功地合成了具有层次结构的方沸石。张海荣等[16]以硅酸钠和硫酸铝为硅铝源,氢氧化钠为碱源,将一定质量的原料与少量水混合直接研磨至黏稠状形成极浓体系,加入晶种和有机模板剂(乙二胺),200 ℃下晶化24 h,晶化产物经550 ℃焙烧脱除有机胺合成了以立方晶系为主的方沸石产品,其对甲醇转化制烯烃的最大转化率为68.2%,但该合成方法存在晶化时间较长、晶化温度高、需要添加晶种和有机模板剂等弊端。
为解决传统水热法中存在的晶化温度高、晶化时间长、产品粒径大、母液难处理等问题,本文提出了一种在极浓体系中以液固相双向转化过程为基础的快速合成小颗粒方沸石的方法(即类固相法[17]),并详细探讨了不同晶化时间、晶化温度、n(Na2O)/n(SiO2)和n(H2O)/n(SiO2)对方沸石合成的影响。
2 实 验
2.1 原料及试剂
水玻璃(分析纯,凯玛特(天津)化工科技有限公司),硫酸铝(分析纯,天津市风船化学试剂科技有限公司),氢氧化钠(分析纯,利安隆博华(天津)医药化学有限公司),氢氧化铝(分析纯,天津市江天化工技术有限公司),去离子水。
2.2 方沸石的制备
2.2.1 类固相法合成方沸石
(1) 硅铝干凝胶的制备:按n(SiO2)/n(Al2O3)=5,称取一定质量的Na2SiO3·9H2O 和Al2(SO4)3·18H2O置于烧杯中,加适量水溶解,加热至60 ℃充分搅拌,混合均匀后冷却至室温,过滤并多次洗涤直至洗涤液中无SO42-存在,再置于100 ℃恒温干燥箱中烘干3 h 得硅铝干凝胶。
(2) 方沸石的合成:将上述硅铝干凝胶放入带有聚四氟乙烯内衬的不锈钢晶化釜中,按一定比例向其中补加氢氧化钠和水,充分搅拌混合均匀,在一定温度下静态晶化一段时间,晶化完成后取出抽滤,固相产物经去离子水洗涤、烘干即可得到方沸石成品。
2.2.2 水热合成法合成方沸石
按n(SiO2):n(Na2O):n(Al2O3):n(H2O)=8:8:1:640,称取一定量的Na2SiO3·9H2O 和Al(OH)3置于三口烧瓶中,加适量水溶解,并在50 ℃恒温水浴中老化2 h。将混合液转移至带聚四氟乙烯内衬的不锈钢高压晶化釜,并置于150 ℃恒温干燥箱中静态晶化24 h,晶化完成后过滤分离固相产物,洗涤、烘干即可得到方沸石。
2.3 表征及分析方法
2.3.1 溶液组分含量分析
采用GB/T 5484-2012 石膏化学分析方法和GB/T 4209-2008 工业硅酸钠滴定法测定母液中铝元素(以Al2O3计)和硅元素(以SiO2计)的含量。
2.3.2 X 射线衍射分析(XRD) 采用德国Bruker 射线衍射仪分析粉体的物相组成,Cu Kα 辐射,管电压40 kV,管电流200 mA,分辨率0.02°,扫描速度为12 °·min-1,扫描范围为5°~90°。
2.3.3 扫描电镜分析(SEM)
采用JSM6700F 型冷场发射扫描电镜观察样品的表观形貌。仪器的分辨率为3.5 nm,放大倍数为15~2.0×105倍。
2.3.4 红外光谱分析(FRIR)
采用德国Bruker 公司生产的113 型红外光谱仪。仪器的各项参数为:扫描范围4 000~400 cm-1,分辨率为4 cm-1,扫描次数为32 次,干燥的样品与KBr 混合比例为1:100。
2.3.5 激光粒度分析 采用美国马尔文仪器有限公司生产的Mastersizer2000 型激光粒度仪测定样品的粒径分布。仪器采用标准操作规程(SOP),粒度范围为0.02~2 000 μm,扫描速度为1 000 次·s-1。
2.3.6 氮吸附-脱附实验分析(BET)
采用美国麦克仪器公司生产的MicromeriticsASAP2020 型物理吸附仪测定样品的比表面积、孔结构和孔径。通过Brunauer-Emmett-Teller (BET)方法测定样品的总表面积。通过应用于等温线吸附分支的Barrett- Joyner-Halenda (BJH)模型获得样品的孔径分布。
2.3.7 方沸石产品的氟离子吸附性能测试
将方沸石产品置于450 ℃的马弗炉中焙烧2.5 h,再用2. 0 mol·L-1的氯化铝溶液浸泡24 h,干燥后再在450 ℃温度下焙烧2.5 h,随炉冷却后备用。将1.5 g 完成改性的方沸石产品投入50 mL 5 mg·L-1氟化钾溶液,室温条件下机械搅拌2 h,用氟离子选择电极法测定上清液中F-浓度,并计算F-去除率。
3 实验结果与讨论
3.1 硅铝干凝胶的表征
硅铝干凝胶的XRD 谱图和SEM 图分别如图1 和2 所示,其XRD 谱图中未出现明显的特征衍射峰,且SEM 图中形貌杂乱无章,说明硅铝干凝胶为无定型态物质。利用2.3.1 节测定方法确定凝胶洗涤液的组成,经质量核算可得硅铝干凝胶中各组分的摩尔比约为n(SiO2):n(A12O3):n(H2O)=15:3:11,该组成与原料中的硅铝摩尔组成n(SiO2)/n(Al2O3)=5 保持一致。

图1 硅铝干凝胶的XRD 谱图 Fig.1 XRD pattern of silica-alumina xerogel

图2 硅铝干凝胶的SEM 图 Fig.2 SEM image of silica-alumina xerogel
3.2 不同条件对方沸石合成的影响
合成方沸石所选用的硅源和铝源为硅铝干凝胶,晶化过程中凝胶相发生溶解、成核、生长等过程,由于其组成n(SiO2)/n(Al2O3)=5 已固定,故文中n(SiO2)/n(Al2O3)对方沸石合成过程的影响未做讨论,仅讨论了晶化时间、晶化温度、n(Na2O)/n(SiO2)和n(H2O)/n(SiO2) 这4 个因素对方沸石合成的影响。
3.2.1 晶化时间对合成方沸石的影响
晶化时间与体系中的碱浓度、含水量、晶化温度等因素密切相关。当晶化温度T=150 ℃,n(SiO2)/n(Al2O3)=5,n(Na2O)/n(SiO2) = 0.26,n(H2O)/n(SiO2) = 11.10 时,不同晶化时间时合成方沸石产品的XRD 谱图和SEM 图分别如图3 和4 所示。

图3 不同晶化时间所得产品的XRD 谱图 Fig.3 XRD patterns of synthesized samples at different crystallizing times

分析图3、4 可得,反应时间为2 h 时,产品的XRD 谱图中主要出现八面沸石、P 型分子筛以及部分杂质的特征衍射峰,但峰型不完整且峰强较低,由于八面沸石与方沸石结构上同属于立方晶系[18],晶体结构相似,八面沸石的形成可能是方沸石合成过程的一个初期阶段。晶化时间延长至4 h 时,产品的特征衍射峰主要以P 型分子筛为主,产品形貌表现为粒径均匀的粗糙小球。以上结果可能是由于晶化反应前期主要发生硅铝凝胶的溶解过程,虽然高碱浓度可以加速凝胶溶解,缩短诱导期和成核期,但短时间内并不能合成完整的方沸石晶体。当晶化时间延长为6 h 时,P 型分子筛逐渐完成了向方沸石的相变转化,此过程进一步说明P 型分子筛结构较不稳定。此时XRD 谱图中虽出现了明显的方沸石特征衍射峰,但仍有少量的杂质峰存在,SEM 图也验证了这一事实。继续延长晶化时间,结果表明晶化6~10 h 均可合成方沸石且其特征衍射峰的峰型均较完整,峰强逐渐增强。晶化时间为6~10 h 时,体系中自由水不断被消耗,凝胶相的溶解受到限制,导致部分凝胶相未经溶解即直接在固相环境中发生浓缩重排、成核和晶体生长等过程[17],同时部分凝胶相依托前期液相转化形成的晶核完成快速生长,使得整个晶化周期明显缩短。但对于整个晶化过程而言,晶化时间过长又会导致液相体系中自由水消耗增加,使液相转化过程受阻。
固相转化过程也因缺少晶核而受到限制,使得部分原料无法完全参与晶化过程,致使产品结晶度降低,文中晶化时间为10 h 的实验结果也证实了这一点。结合图4(d)可以发现,晶化时间为8 h 时,合成产品的杂质最少,粒度均匀,晶型结构清晰,故8 h 为该方法的适宜晶化时间。
3.2.2 晶化温度对合成方沸石的影响
晶化温度是影响沸石形成的一个重要因素。当晶化时间t = 8 h,n(SiO2)/n(Al2O3)=5,n(Na2O)/ n(SiO2) = 0.26,n(H2O)/n(SiO2) = 11.10,不同晶化温度时合成方沸石产品的XRD 谱图和SEM 图分别如图5 和6所示。
晶化温度的变化会影响体系内水的自生压,影响凝胶相的溶解、晶核的形成、晶体的生长以及介稳态间的相变等过程。分析图5、6 可知,晶化温度为130 ℃时,产品的XRD 谱图中呈现的特征衍射峰以P 型分子筛为主,虽然出现少量方沸石的特征衍射峰但强度较弱,其SEM 图也主要呈现为粗糙球状的P型分子筛。这说明当水汽共存的晶化体系温度较低时,用于溶解凝胶的自由水相对较多有利于凝胶相的溶解再结晶转化为沸石结构[19];但温度较低时凝胶相溶解度较低,又有利于固相转化直接形成沸石结构。通常水热法合成的分子筛产品形貌均较规整,而图6(a)中P 型分子筛的形貌表面粗糙,说明固相转化体系因原料分布不均匀容易造成沸石表面粗糙,所以该温度下主要经固相转化形成了P 型分子筛。继续升高温度到140 和150 ℃时,XRD 谱图中方沸石产品的特征衍射峰峰型趋于完整且峰强逐渐增强,其中晶化温度为150 ℃时,方沸石的晶型结构最为清晰完整,粒径均匀。这说明升高温度可促进凝胶相溶解过程,有利于凝胶相的溶解再结晶转化为方沸石结构。当晶化温度为170 和190 ℃时,方沸石的特征衍射峰强度逐渐降低且出现了水合硅酸铝酸钠的杂质峰,方沸石晶型遭到破坏,晶粒表面出现明显裂痕。此结果说明体系升高温度虽可增大凝胶相的溶解度,但温度过高又会因水的汽化程度过高使体系的自由水明显减少而限制凝胶溶解,不利于液相成核过程;同时固相转化过程也会因液相过程无法继续为其提供晶核而受阻,此时固相转化和液相转化过程受到双重限制,不利于方沸石的合成,故方沸石合成体系的晶化温度不宜过高。由上可见,150 ℃为该方法适宜的晶化温度。
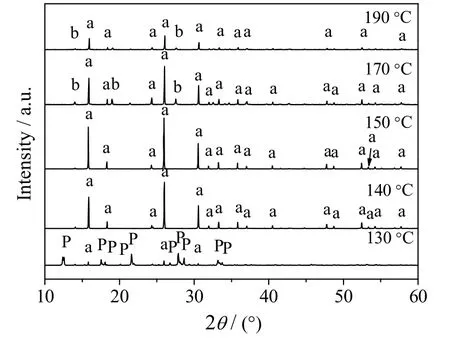
图5 不同晶化温度下所得产品的XRD 谱图 Fig.5 XRD patterns of synthesized samples at different crystallizing temperatures

3.2.3 n(Na2O)/n(SiO2)对合成方沸石的影响
沸石合成是在碱性或强碱性条件下进行,体系碱浓度会影响合成过程的晶化速率和产品形貌。当晶化时间t = 8 h,晶化温度T = 150 ℃,n(SiO2)/n(Al2O3) = 5,n(H2O)/n(SiO2) = 11.10 时,不同n(Na2O)/n(SiO2)配比时合成方沸石产品的XRD 谱图和SEM 图分别如图7 和8 所示。
分析图7 可得,当反应体系中n(Na2O)/n(SiO2)比值处于0.22~0.34 时,产品XRD 谱图的相同位置均出现了方沸石的特征衍射峰,故n(Na2O)/n(SiO2)在此范围内均能合成方沸石产品。如图8(a)所示,当n(Na2O)/n(SiO2)=0.22 时,方沸石产品的粒径不均匀、晶体结构特点不清晰。沸石合成体系的碱浓度通常用OH-/Si(摩尔比,下同)的大小来表示,适宜浓度的OH-会破坏凝胶相中硅铝酸盐的化学键,促进铝酸根离子发生水解、解聚和重排反应,改变体系中原料的聚合态及分布情况,加快晶化速率,缩短诱导期和成核期,而n(Na2O)/n(SiO2) = 0.22 时体系碱浓度较低,不利于完成上述过程,故不能合成结晶良好的方沸石产品。当n(Na2O)/n(SiO2)增大至0.26,方沸石产品特征衍射峰强度明显增强,晶体结构清晰完整,粒径均匀。这说明适当提高碱浓度可加快硅、铝缩聚反应,促进晶体生长,改变晶体形貌,有利于方沸石(富硅沸石)的形成[20],同时钠离子在反应过程中可发挥“桥连”作用,使晶型规则有序。继续增大n(Na2O)/n(SiO2)超过0.26 时,XRD 谱图中方沸石的特征衍射峰强度逐渐减弱,并出现了硅铝酸钠水合物等杂质峰。观察SEM 图也验证了这一点,与图8(b)相比,图8(c)、(d)产物中无定型杂质含量明显增多,晶体形成聚合态结构且粘连程度逐渐增大,不再呈现明显的单一晶型结构。由上可见,n(Na2O)/n(SiO2) = 0.26 为该方法适宜的碱硅比。

图7 不同n(Na2O)/n(SiO2)时所得产品的XRD 谱图 Fig.7 XRD patterns of synthesized samples at different n(Na2O)/n(SiO2) ratios

图8 不同n(Na2O)/n(SiO2)时所得产品的SEM 图 Fig.8 SEM images of synthesized samples at different n(Na2O)/n(SiO2) ratios
3.2.4 n(H2O)/n(SiO2)对合成方沸石的影响
作为分子筛的合成过程的关键因素,水不仅是晶化过程的反应介质,还作为反应物参与分子筛的形成。当晶化时间t =8 h,晶化温度 T = 150 ℃,n(SiO2)/n(Al2O3) = 5,n(Na2O)/n(SiO2) = 0.26 时,不同n(H2O)/n(SiO2)时合成方沸石产品的XRD 谱图和SEM 图分别如图9 和10 所示。
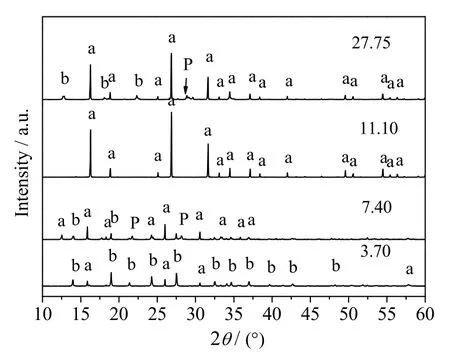
图9 不同n(H2O)/n(SiO2)时所得产品的XRD 谱图Fig.9 XRD patterns of synthesized samples at different n(H2O)/n(SiO2) ratios

图10 不同n(H2O)/n(SiO2)时合成产品的SEM 图 Fig.10 SEM images of synthesized samples at different n(H2O)/n(SiO2) ratios
分析图9、10 可得,当n(H2O)/n(SiO2) = 3.70 时,XRD谱图中仅在2θ = 16.0o、26.0o和31.0o处出现方沸石的特征衍射峰,其余特征峰主要为水合硅酸铝钠的特征衍射峰,其SEM图显示产物中有未完全成型的方沸石晶体出现,同时也有部分杂乱无章小颗粒或颗粒聚合体存在。虽然此时体系中含水量很少,但对方沸石形貌和结晶速率的影响远大于水热合成过程,这是由其参与分子筛合成机理决定的[21],n(H2O)/n(SiO2) = 3.70 时,体系中含水量很少且高温条件下主要以水蒸气形式存在,由于饱和水蒸气的传质和传热效果明显低于液态溶剂,使得钠、硅铝酸根等离子无法均匀混合,进而造成产物的结晶度较低。n(H2O)/n(SiO2)增大至7.40 时,方沸石的特征峰峰强逐渐增强,但仍有少量水合硅酸铝钠和P 型分子筛的特征峰存在,方沸石的晶体结构趋于清晰完整,但粒径不均。n(H2O)/n(SiO2) 增大至11.10 时,方沸石的特征衍射峰趋于规整且峰强度达到最大值,此时晶体结构清晰完整,粒径均匀。此时体系中适宜的含水量既保证了凝胶相的充分溶解,且高离子浓度又有利于大量晶核的快速形成;同时低含水量可以很大程度地抑制硅铝物种的迁移,则可溶性物相只能通过少量水汽凝结局部润湿而成晶,有利于小粒径的方沸石的形成[22]。此外,高压水蒸气还可以充当造孔剂使方沸石形成较大的比表面积和良好的孔结构。当n(H2O)/n(SiO2) = 27.75 时,产品的XRD 谱图中虽然呈现出较为完整的方沸石特征衍射峰,但同时有较多杂质峰出现,此时产品形貌以空心球壳和规整方沸石晶体为主,说明该合成体系含水量过大不利于方沸石晶体的形成。由上可见,n(H2O)/n(SiO2) = 11.10 为该方法适宜的水硅比。
综上所述,可以确定类固相法合成小粒径方沸石的适宜条件为:晶化时间为8 h,晶化温度为150 ℃,硅铝干凝胶的摩尔组成为n(SiO2)/n(Al2O3) = 5,初始合成原料摩尔组成n(Na2O)/n(SiO2) = 0.26,n(H2O)/ n(SiO2) = 11.10。
3.3 方沸石的结构和性能测试
3.3.1 方沸石产品的XRD 和SEM 分析
采用类固相法(用S 表示,下同)和水热法(用H 表示,下同)制备样品的XRD 谱图和SEM 图如图11 和12 所示,XRD 谱图中在2θ = 15.8°、18.5°、24.5°、26.0°、30.5°、32.0°、33.3°、35.8°、48.8°和52.5°等处均出现了方沸石的特征衍射峰,说明2 种方法均能合成纯度较高、结晶良好的方沸石产品,2 种方法合成的产品形貌均为典型的晶体结构,其中类固相法合成的方沸石粒径均匀、结构单一,而水热法合成的方沸石晶粒整体较大、大小不一、且多以聚合体形式存在。
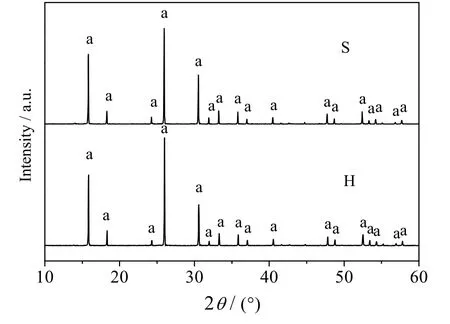
图11 不同合成方法所得产品的XRD 谱图 Fig.11 XRD patterns of synthesized samples by different synthetic methods

图12 不同合成方法所得产品的SEM 图 Fig.12 SEM images of synthesized samples by different synthetic methods
3.3.2 合成产品的元素含量分析
合成产品的EDS 表征数据如图13 和表1 所示,分析数据可得2 种方法合成的方沸石产品的元素组成比较接近,基本接近方沸石(Na[A1Si2O6]·H2O)的理论摩尔组成n(Na):n(Al):n(Si):n(O) = 1:1:2:6。

图13 不同合成方法所得产品的EDS 谱图 Fig.13 EDS i mages of synthesized samples by different synthetic methods
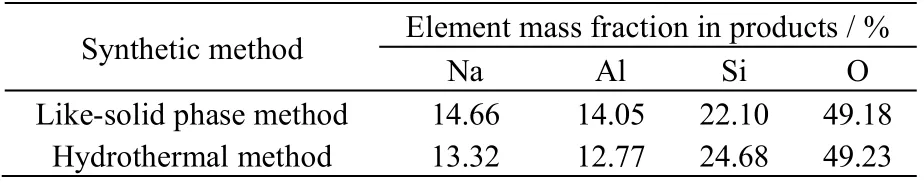
表1 方沸石产品中各元素的质量分数 Table 1 Elemental mass fractions in the analcite products
3.3.3 方沸石产品的FRIR 分析
采用溴化钾压片法进行测试,可以得到合成方沸石样品在400~4 000 cm-1的红外光谱结果,如图14 所示。图中3 600 cm-1为O-H 羟基伸缩振动吸收峰,1 640 cm-1处为吸附水弯曲振动峰,说明合成方沸石具有亲水性,1 000 cm-1附近为二氧化硅中Si-O 的伸缩振动吸收峰500~750 cm-1处为四面体Si-O-Si 或Al-O-Al 的对称伸缩振动峰[2],以上红外光谱图和文献[23]报道的方沸石红外光谱图相似,说明类固相法和水热法均能合成出结晶良好的方沸石产品。

图14 不同合成方法所得产品的红外谱图Fig.14 IR spectra of synthesized samples by different synthetic methods
3.3.4 方沸石产品的粒径分析
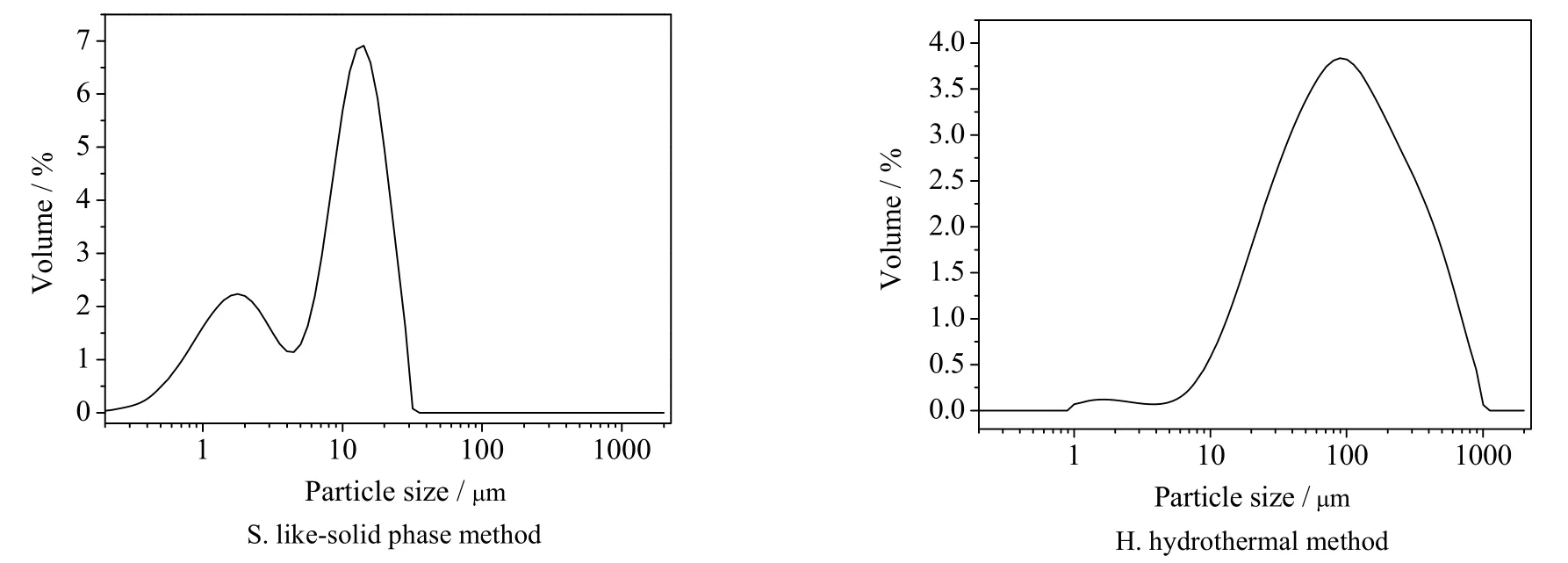
图15 不同合成方法所得产品的粒径分布图Fig.15 Size distribution of synthesized samples by different synthetic methods

表2 方沸石产品的粒径分布 Table 2 Size distribution of the analcite products
方沸石产品的粒径通过激光粒度仪分析可得图15 和表2 数据,类固相法合成的方沸石产品粒径分布90% 以上集中在0.1~20 μm,其粒径分布主要集中在10 μm 左右;而水热法合成的产品粒径分布90% 以上集中在1~370 μm,粒径分布主要集中 在90 μm 左右,其中大粒径产品的形成主要由于水热合成法的部分产品晶面间相互嵌连、紧密排列造成产品板结,若采用机械方法(如超声清洗、研磨等)消除板结现象会破坏方沸石的形貌,故测试前未进行消除板结处理。对比以上数据可得,类固相法可以合成粒径较小的方沸石产品。
3.3.5 方沸石产品的结构和除氟性能分析
方沸石产品的比结构特征和除氟性能数据如表3 所示。与采用传统水热法合成的大粒径方沸石产品相比,采用类固相法合成的小粒径方沸石产品的比表面积、孔体积和孔径均有所增大,且对等浓度的氟化钾溶液,其除氟率提高约16.1%,由此说明类固相法合成的小颗粒方沸石产品具有更高的氟离子吸附性能。

表3 方沸石产品的结构特征和除氟性能 Table 3 Structural characteristics and fluoride removal performance of the analcite products
4 结 论
(1) 采用类固相法以硅铝干凝胶为硅源和铝源,氢氧化钠为钠源和碱源,按照以下适宜合成条件:晶化时间8 h,晶化温度150 ℃,凝胶摩尔组成n(SiO2)/n(Al2O3) = 5,原料摩尔组成n(Na2O)/n(SiO2) = 0.26,n(H2O)/n(SiO2) = 11.10,可以合成出结晶良好、粒径均匀的小颗粒方沸石产品。
(2) 与传统水热合成方法相比,类固相法合成方沸石的工艺具有晶化时间短、原料利用率高、反应耗水少、产品粒径小等特点,有效地解决了传统水热法生产周期长、母液含水量大以及母液处理难等问题。
(3) 与传统水热法合成的方沸石产品相比,类固相法合成的小粒径方沸石产品具有更大的比表面积、孔体积、孔径以及更高的氟离子吸附性能。
