成人散发型神经元核内包涵体病1例报告
2020-09-15欧阳桂兰朱海兵谢高生钟善全
欧阳桂兰 朱海兵 谢高生钟善全
神经元核内包涵体病 (neuronal intranuclear inclusion disease,NIID)是一种慢性进展的神经变性疾病,其病理特征是中枢、周围神经系统以及内脏器官中的嗜酸性透明核内包涵体形成[1]。因其临床表现多变而被认为是一种异质性疾病,也给临床带来诊断困难[2]。近年来发现皮肤活检对NIID的诊断帮助,该病的诊断数量逐渐增加。为加深对该病的进一步认识,现分析一例经皮肤活检及基因检测确诊的NIID患者,以探讨该病的临床、影像学特点及诊断方法。
1 临床资料
患者男,63岁,右利手,因“小便失禁3年,认知功能下降2年,加重2个月”于2019年8月5日入院。患者于3年前开始出现小便失禁,小便不能控制,容易尿在身上。2年前出现记忆力下降,容易丢三落四,伴有头晕,四肢麻木、乏力,行走稍迟缓及四肢轻微颤动,但生活能自理。上述症状逐渐进展,并且外出容易迷路。2019年6月始症状较前有所加重,故来我院就诊。1年前诊断患有糖尿病,服用“阿卡波糖片”控制血糖,血糖控制基本理想。既往有吸烟史30年余,每日20支;有少量饮酒史30年余,每日约50~100 mL白酒。入院时神经系统体检:卧位血压为105 mmHg/75 mmHg(1mmHg=0.133kPa),立位血压第 1 分钟 102 mmHg/70mmHg,第2分钟108mmHg/78mmHg,第 3分钟112mmHg/76 mmHg;记忆力、计算力减退,简易精神智能量表评分为23分(初中文化);双侧瞳孔无缩小,左:右=3.0 mm:3.0 mm,对光反射正常,无眼震;双下肢膝关节以下浅感觉稍减退,振动觉减弱;四肢肢体远近端肌力4级,四肢腱反射减弱;双手轮替试验稍差,双侧指鼻试验基本准确,双侧跟膝经试验稍差,闭目难立征睁闭眼试验阴性。双侧病理征未引出。余神经系统检查未见阳性体征。实验室检查:血常规,肝肾功能、血脂、电解质等血生化,风湿免疫项目,铜蓝蛋白,甲状腺功能及肿瘤标志物等均正常;艾滋病病毒抗体、梅毒螺旋体抗体均阴性;空腹血糖7.96 mmol/L,糖化血红蛋白10.4%。脑脊液常规正常,生化示脑脊液总蛋白958 mg/L(正常值150~450 mg/L),余未见异常。脑电图示边缘性脑电图。肌电图示多发性周围神经损害,左尺神经未引出感觉神经动作电位,右尺神经、右正中神经、左腓肠神经感觉神经波幅减低,右正中神经感觉传导速度减慢,右尺神经感觉传导速度正常低限;左腓总神经运动神经波幅减低,传导速度减慢;左正中神经及右胫神经、腓总神经运动传导速度减慢,波幅未见明显异常;双足皮肤交感反应异常。2019年8月6日我院头颅磁共振示:双侧额叶白质区T2加权成像(T2weighted imaging,T2WI)高信号;双侧额叶皮质-髓质交界区弥散加权成像 (diffusion weighted imaging,DWI)高信号;双侧小脑蚓部旁液体衰减反转恢复(fluidattenuated inversion recovery,FLAIR)序列呈片状高信号,左侧小脑中脚FLAIR序列呈片状稍高信号,小脑萎缩;磁共振波谱成像(MRS)示胆碱(Cho)、肌酸(Cr)、天冬酰胺(NAA)峰值下降不明显,NAA/Cr比值无下降(图1 A~E)。2019年9月5日皮肤肌肉病理及基因报告:皮肤肌肉病理报告示汗腺导管上皮细胞和成纤维细胞核内出现嗜酸性物质沉积,该物质p62阳性,电镜进一步证实存在核内包涵体,符合NIID在皮肤病理的改变特点(图2 A~C);基因报告示患者血标本经过RP-PCR检测NOTCH2NLC基因,在其5UTR区域存在超过100次的GGC突变,提示NIID致病基因阳性。诊断:①神经元核内包涵体病;②2型糖尿病。
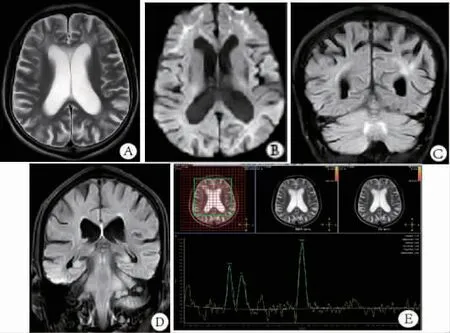
图1 头颅影像 双侧额叶白质区T2WI高信号(A);双侧额叶皮质-髓质交界区DWI高信号(B);双侧小脑半球内侧FLAIR序列呈片状高信号(C);左侧小脑中脚FLAIR序列呈片状稍高信号,小脑萎缩 (D);Cho、Cr、NAA 峰值下降不明显,NAA/Cr比值无下降(E)。
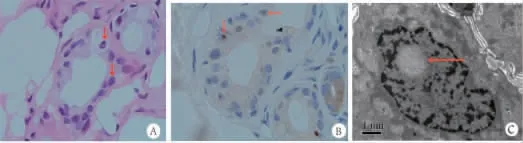
图2 皮肤病理结果 苏木精一伊红(HE)染色可见皮肤汗腺导管上皮细胞内嗜酸性透明核内包涵体(A);抗P62抗体阳性(B);电镜皮肤成纤维细胞核内可见球状包涵体(C)。
2 讨论
NIID也称为神经元核内透明包涵体病,是一种少见的慢性神经变性疾病。1968年,LINDENBERG等[2]经脑组织病理活检证实的第1例儿童期起病的28岁男性NIID患者,以精神发育迟滞、共济失调、进行性痉挛为主要症状。随后在1980年,SUNG等[3]诊断1例21岁女性NIID患者。截止到2011年,世界上仅报告约40例[1]。2011年日本学者SONE通过皮肤活检确诊该病,此后日本学者报告该病超过60例。
NIID的临床特征多种多样,存在高度异质性,多为慢性或亚急性起病,可表现为缓慢进展的中枢神经、周围神经及自主神经等症状,也可表现为亚急性起病的脑病症状,或为反复发作的急性病程。病程1~44年不等,发病年龄从婴儿到老年人均可见,男女之比为1:2,分为儿童型、少年型及成人型[4]。儿童型、少年型患者多以共济失调或精神发育迟滞、行为异常为首发症状,而成年型患者早期症状多为肢体无力或认知功能障碍。此外,该病也可合并有狼疮肾炎[5]、蛋白尿[6]、糖尿病及假性肠梗阻等非神经系统症状。根据是否家族遗传而分为家族型、散发型,而根据临床症状分为肢体无力型(多为下肢)、痴呆型。国内外研究发现,散发型病例多以痴呆为首发症状,而在家族型病例中以肢体无力与痴呆起病比例相当;成人型NIID以肢体无力起病的患者,在病程发展20年后可出现痴呆,而以痴呆为起病症状者,肢体无力相对少见[1]。通过国内外的病例研究报道[1,7-8],成人型NIID多在50岁后起病,少数病例起病年龄较早,临床症状主要为:①中枢神经症状:痴呆、精神行为异常、共济失调、痫性发作、发作性意识障碍、卒中样发作、脑炎样症状、震颤、强直;②外周神经症状:肢体无力、感觉障碍;③自主神经症状:尿便障碍、瞳孔变小、晕厥、呕吐。我们报道的该病例患者60岁起病,病程3年,临床症状主要为痴呆、肢体无力、感觉障碍及尿便障碍等。
2014年,SONE等[9]报道了经皮肤病理确诊的3例成人型NIID,发现其临床症状各异,但均存在大脑皮质-髓质交界区DWI高信号,即提出该征象是NIID诊断的重要线索,是采取皮肤活检的首要条件。2016年,SONE等[1]对57例成人NIID患者的研究发现,81.8%的家族型和100%的散发型患者均有此征象。大脑皮质-髓质交界区DWI高信号可能与核内异常蛋白的堆积和蛋白降解系统的功能障碍有关。2019年,我国学者LIU等[8]行多种功能影像检查1例NIID患者发现,除DWI典型征象外,还可见NAA/Cr值下降、白质纤维束受损及大脑皮质葡萄糖代谢降低,这些功能成像结果改变可能与认知功能下降有关。所以,大脑皮质-髓质交界区的DWI高信号是NIID的特征性征象。国内学者陈为安等[7]将该征象取名为皮质下绸带征,如有此征象可高度怀疑NIID。此外,也可见双侧脑白质弥漫对称T2FLAIR序列高信号。国外学者SUGIYAMA等[10]观察8例成人NIID小脑磁共振影像发现,8例患者小脑萎缩,6例患者小脑半球内侧及4例患者小脑中脚FLAIR高信号,认为该征象也是NIID的诊断指标。我们报道的此病例患者,除在大脑皮质-髓质交界区出现条带状DWI高信号,也发现双侧小脑半球内侧及左小脑中脚FLAIR片状高信号,但MRS中NAA/Cr值无下降。
在2011年之前,主要通过腓肠神经、直肠、尸体活检确诊NIID。2011年,SONE等[11]发现皮肤汗腺细胞核内包涵体病理学特点与神经元核内包涵体一致,从而认为皮肤活检是确诊NIID的可靠手段。NIID组织病理学特点为,HE染色呈嗜酸性透明包涵体,免疫组织化学可见抗泛素抗体和抗p62抗体阳性,在电镜下可见包涵体为无膜结构的纤维物质组成[1]。近年来,国外学者研究认为NOTCH2NLC基因的GGC突变重复扩增致神经元毒性的可能,是NIID的病因[12-13]。此外,GGC重复扩增已被确定为中国人群NIID发病的遗传病因[14]。我们报道的此病例经皮肤病理活检确诊,也发现在NOTCH2NLC基因的5UTR区域GGC突变重复扩增超过100次。
此外,神经电生理检查也可发现胫神经、腓总神经及正中神经受累常见,表现为感觉神经及运动神经传导速度减慢,而波幅降低在肢体无力型患者中相对多见。有癫痫发作的患者可见痫样放电的脑电图[15]。脑脊液检查特异性不高,多数患者脑脊液蛋白轻度升高,但也有完全正常[1]。我们报道此病例患者神经电生理检查发现类似异常表现及脑脊液蛋白升高。
NIID需要与脆性X染色体综合征相鉴别,后者在临床中多表现为智能发育迟缓及锥体外系等症状,也有存在脑白质病变的病例报道,并且在DWI序列上呈现灰白质交界区高信号。所以从临床、影像学及病理学检查两者难以区别,在诊断NIID中需要完善CGG片段扩增检测以除外脆性X染色体综合征。当然,也需要与代谢性脑病、脊髓小脑共济失调、帕金森病及多系统萎缩等疾病相鉴别。
NIID目前无特异性治疗方法,主要以对症支持治疗为主。如以亚急性脑炎为主要表现的患者,可考虑短期糖皮质激素冲击治疗减轻水肿及改善意识状态,但长期预后未知[1]。也有个别病例报道以反复脑炎发作的患者,予以神经营养及补充B族维生素、维生素C等治疗后,短期内症状均得到可逆效果[16]。
综上所述,大脑皮质-髓质交界区DWI高信号与小脑半球内侧、小脑中脚FLAIR高信号是NIID诊断线索;皮肤病理活检及基因学检查有助确诊。
